今天为大家介绍一篇工作,是二月份发表在Nature上的“分子内二价胶水”的文章。通讯作者是邓迪大学的Alessio Ciulli教授和奥地利科学院分子医学研究中心的Georg E. Winter教授。Ciulli课题组关注蛋白-蛋白相互作用(PPI)中的化学生物学、结构生物学问题,尤其关注小分子化合物对于PPI的调节。Winter课题组关注小分子调控蛋白质降解,尤其是不可成药靶点的降解。故事还要从Indisulam讲起。2017年,磺酰胺类化合物Indisulam登上Science1,它介导CUL4A/BDCAF15与RBM39的互作,导致RMB39被泛素化,最终被降解(图1)。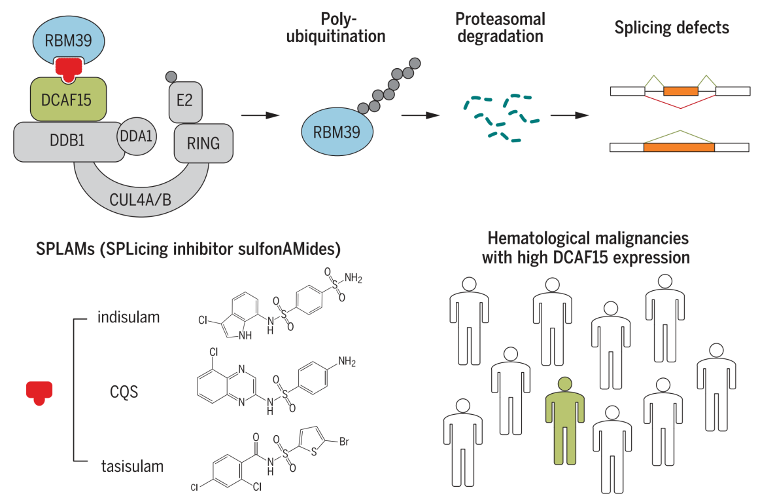
图1. 磺胺类药物诱导RBM39与CUL4A/BDCAF15形成复合物
E7820是Indisulam的类似物,它也可以诱导RBM39与CUL4A/BDCAF15形成复合物(图2)2。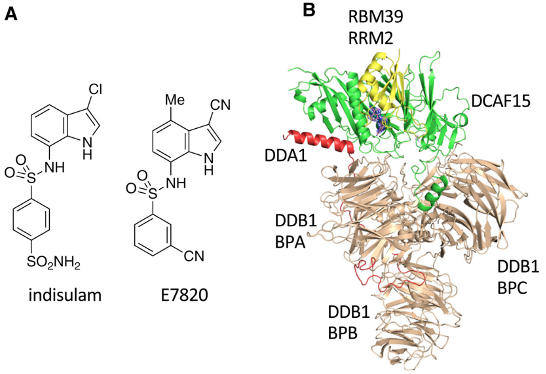
图2. A)Indisulam及E7820结构 B) BM39-E7820-DCAF15复合物结构一些将E7820作为E3连接酶底物受体binder进行PROTAC分子设计的尝试没有取得很好的结果,靶蛋白降解效率很低。但是, E7820与(+)-JQ1(JQ1是BRD4配体,在分子胶设计领域非常常用)通过刚性Linker连接的融合分子——IBG1,活性很强,降解BRD4的DC50达到0.15 nM(图3)。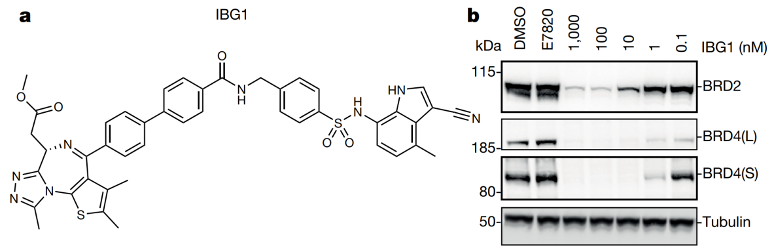
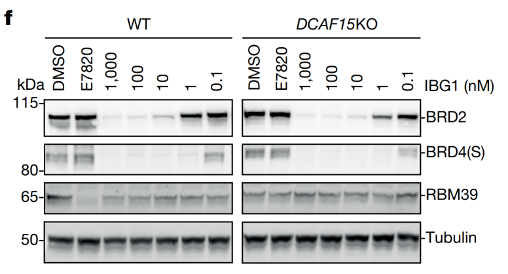
通过FACS-based CRISPR–Cas9 screens,作者发现CRL4DCAF16 复合物中的组分与IBG1给药后BRD4稳定性有较强相关性。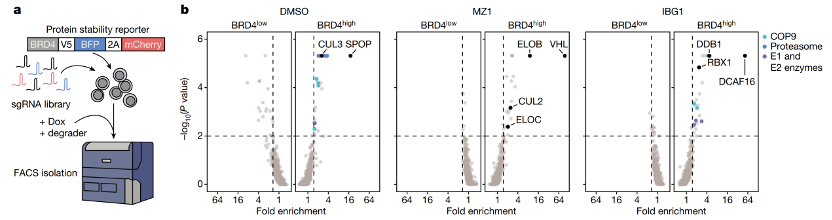
图5. FACS-based CRISPR–Cas9 screens流程示意图作者进一步证明,敲除DCAF16、DDB1消除了IBG1降解BRD4的能力。按照一般的思路,那应该是形成了BRD4-IBG1-DCAF16三元复合物,导致BRD4泛素化,然后被降解。但是E7820衍生物1a,1b,1c,1d并没有拮抗IBG1诱导的BRD4降解(图6A),证明E7820片段不是扮演PROTAC中E3连接酶binder的角色;而JQ1的衍生物1e,1f,1g也没有降解BRD2/4的能力(图6B),说明完整的E7820结构是降解所必需的。综上所述,IBG1分子发挥功能的机制与PROTAC不同。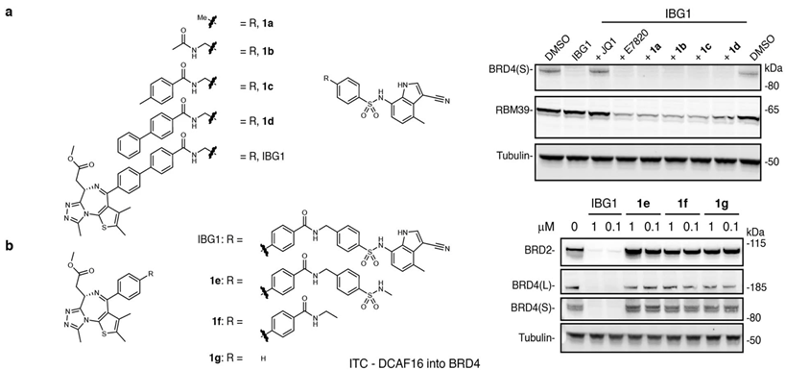
图6. A)E7820衍生物结构 B)JQ1衍生物结构这篇文章最神奇的部分到了,在此之前先看BRD4的结构(图7)。BRD4有两个Bromodomain(BD1和BD2),而JQ1跟这两个结构域都有纳摩尔级的结合力3。以往研究Bromodomain抑制剂及其复合物结构的时候,通常只会用单个的BD 1domain或者BD2 domain。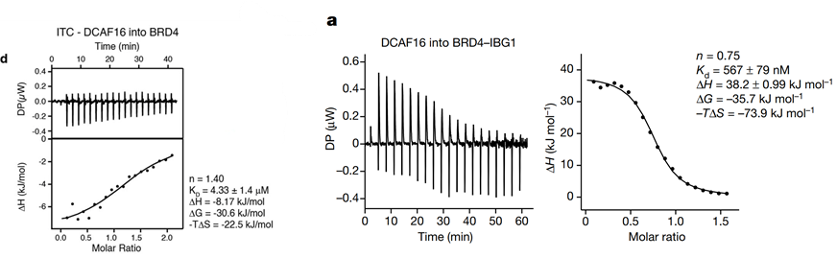
但这篇文章截取了BD1-NLS-BD2这一段进行研究,后文将该截短体称为BRD4Tandem。作者发现DCAF16与BRD4Tandem有着天然的亲和力,IBG1的加入增强了DCAF16-的结合,也改变了结合模式,结合由焓驱动转变为熵驱动(图8)。图8. d)DCAF16滴定BRD4Tandem a)DCAF16滴定BRD4Tandem-IBG1紧接着作者证明了BD1/2同时存在是IBG1发挥作用的基础,也解出了BRD4Tandem-IBG1-DCAF16-DDB1的复合物结构(图9)。尺寸排阻色谱证明IBG1将BRD4Tandem拉紧;电镜结构表明IBG1与BRD4Tandem互作后形成一个新的疏水界面,DCAF16主要通过疏水作用与该界面互作。这也解释了为什么在IBG1存在时,DCAF16与BRD4Tandem的互作由熵驱动。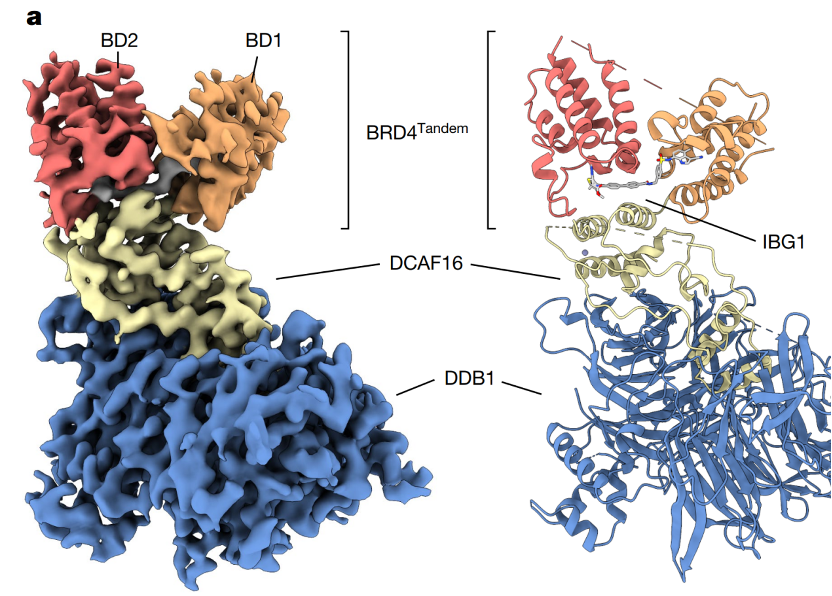
同时,复合物结构也表明JQ1占据了BD2 domain,E7820片段占据了BD1 domain。在此之前也有过磺酰胺结构的Bromodomain抑制剂被报道,将以前结构中的抑制剂与E7820片段叠合,发现他们采取类似的构象(图10)。但磺胺片段与Bromodomain的结合力远弱于JQ1。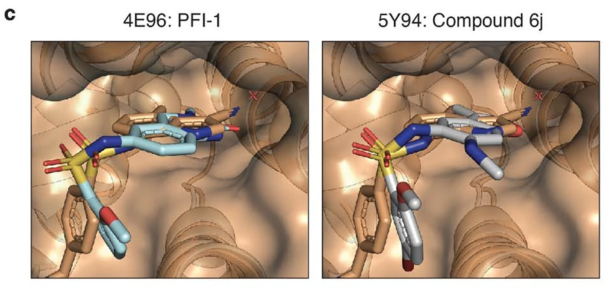
图10. 其他磺胺类抑制剂晶体结构中的pose与本研究中的E7820叠合图,E7820为黄色作者猜想将两个强Bromodomain抑制剂通过类似的方式组装起来,可能会达到更好的效果(图11)。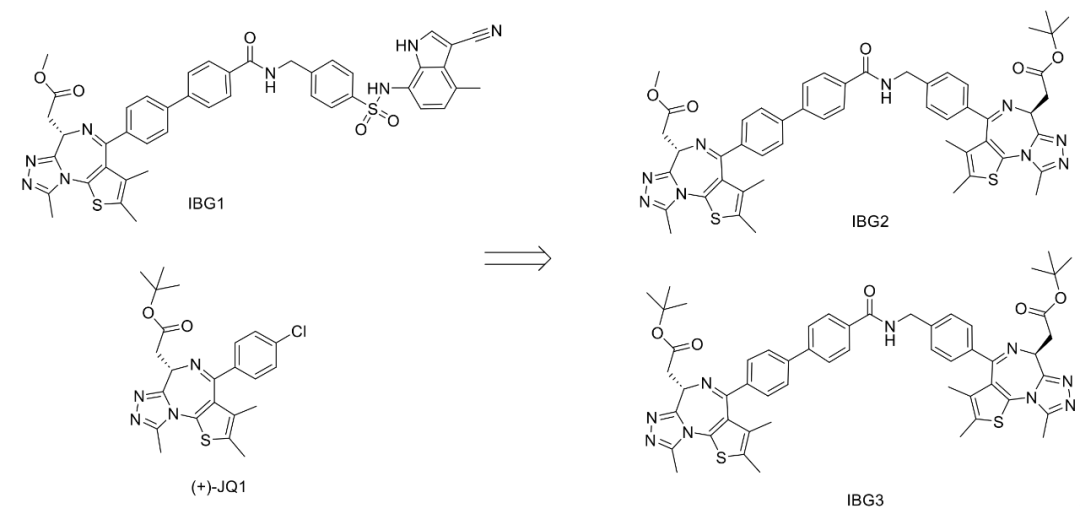
IBG3的活性确实强于原始分子IBG1,降解BRD4的EC50达到了6.7 pM,三元复合物形成的ΔG绝对值更大。但以往也有人做过类似的二价Bromodomain抑制剂(图12),抑制效果很强,但并没有降解BRD4的能力。他们与IBG3的区别主要有三点:1)连接链为柔性,2)连接链与JQ1连接的位置不同,3)连接链长度不同。具体是哪个因素更重要,还需要后续深入地探讨。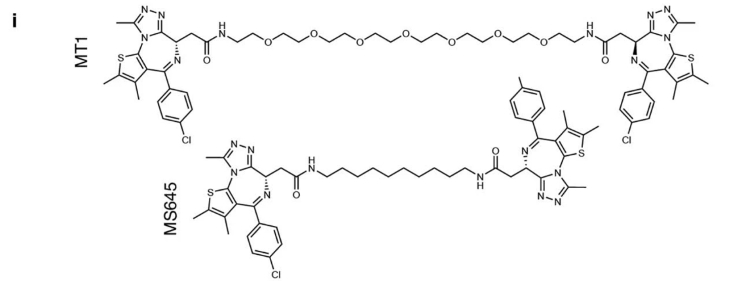
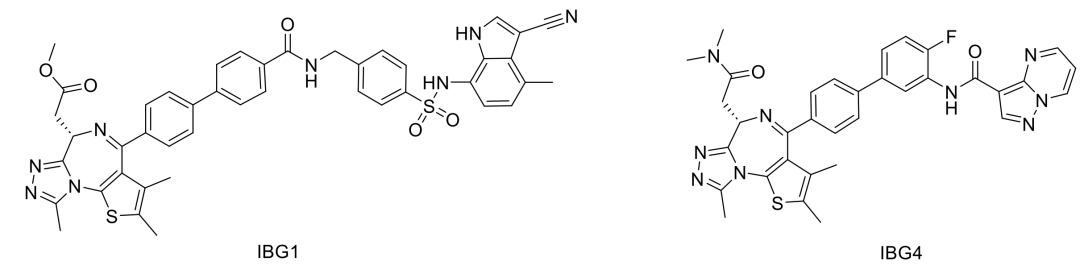
作者在专利中还发现类似的结构——IBG4(图13),该化合物也能降解BRD4,机制与IBG1类似,但是IBG4降解BRD4依赖DCAF11,而不是DCAF16,且DCAF11和16在结构上相似性较小。这篇文章无疑是成功的,他们首次发现分子内二价胶水,拓宽了分子胶类型,并成功理性设计出皮摩尔级的分子内二价胶(IBG1→IBG3)。更难能可贵的是它也带给我们启示和思考,除了VHL/CRBN配体外,慎重选择新出现的binder,如E7820。双功能分子设计看似简单,实则不然。本篇内容就是从DCAF15→DCAF16→DCAF11,过程曲折。关于合理设计分子胶/双功能分子,有人认为蛋白与E3底物受体间的弱结合或许是分子胶必须的,有人则不这么认为,该工作的结果是倾向于前者的。最近分子胶领域的工作喜欢展示自己的分子设计策略5,但这篇文章提醒我们,要严谨的验证自己设想的机制。分子胶理性设计还是一片蓝海!1 Han, T. et al. Anticancer sulfonamides target splicing by inducing RBM39 degradation via recruitment to DCAF15. Science 356 (2017). https://doi.org/10.1126/science.aal3755
2 Du, X. et al. Structural Basis and Kinetic Pathway of RBM39 Recruitment to DCAF15 by a Sulfonamide Molecular Glue E7820. Structure 27, 1625-1633.e1623 (2019). https://doi.org/10.1016/j.str.2019.10.005
3 Filippakopoulos, P. et al. Selective inhibition of BET bromodomains. Nature 468, 1067-1073 (2010). https://doi.org/10.1038/nature09504
4 Weissman, J. D. et al. The intrinsic kinase activity of BRD4 spans its BD2-B-BID domains. J. Biol. Chem. 297 (2021). https://doi.org/10.1016/j.jbc.2021.101326
5 Gourisankar, S. et al. Rewiring cancer drivers to activate apoptosis. Nature 620, 417-425 (2023). https://doi.org/10.1038/s41586-023-06348-2
本文介绍的工作:Hsia, O., Hinterndorfer, M., Cowan, A.D. et al. Targeted protein degradation via intramolecular bivalent glues. Nature 627, 204–211 (2024). https://doi.org/10.1038/s41586-024-07089-6
内容中包含的图片若涉及版权问题,请及时与我们联系删除



评论
沙发等你来抢