
近日,来自普林斯顿大学电气和计算机工程系、斯坦福大学病理系与RVAC Medicines和Zipcode Bio等单位联合开发了一种5′ UTR语言模型(UTR-LM),由王梦迪教授担任通讯作者,褚晏伊博士和于丹博士共同担任第一作者。该模型旨在解码信使RNA(mRNA)分子起始部位的调控区,以预测和改进基因的翻译表达水平。

方法
本研究采用了多源数据集进行模型的预训练和微调。研究团队收集了来自人类、小鼠、大鼠、鸡和斑马鱼的214,349个5′ UTR序列,以及来自人类肌肉组织、PC3前列腺癌细胞系和HEK 293T细胞系的三个独立数据集。此外,研究还涉及了8个合成库,这些库提供了超过200,000个序列,用于深入探索和训练模型。为了进一步验证模型对不同序列长度的适应性和预测准确性,还采用了两种长度范围为25-100bp的独立测试集进行了序列长度的探索和评估。
UTR-LM模型基于transformer架构,通过自监督学习的方式进行预训练,并结合二级结构(SS)和最小自由能(MFE)等监督信息,以增强模型的预测能力。在多个下游任务上进行微调后,该模型能够准确预测mRNA的核糖体负载量(MRL)、翻译效率(TE)、表达水平(EL)、内部核糖体入口位点(IRES)等关键生物学特性。
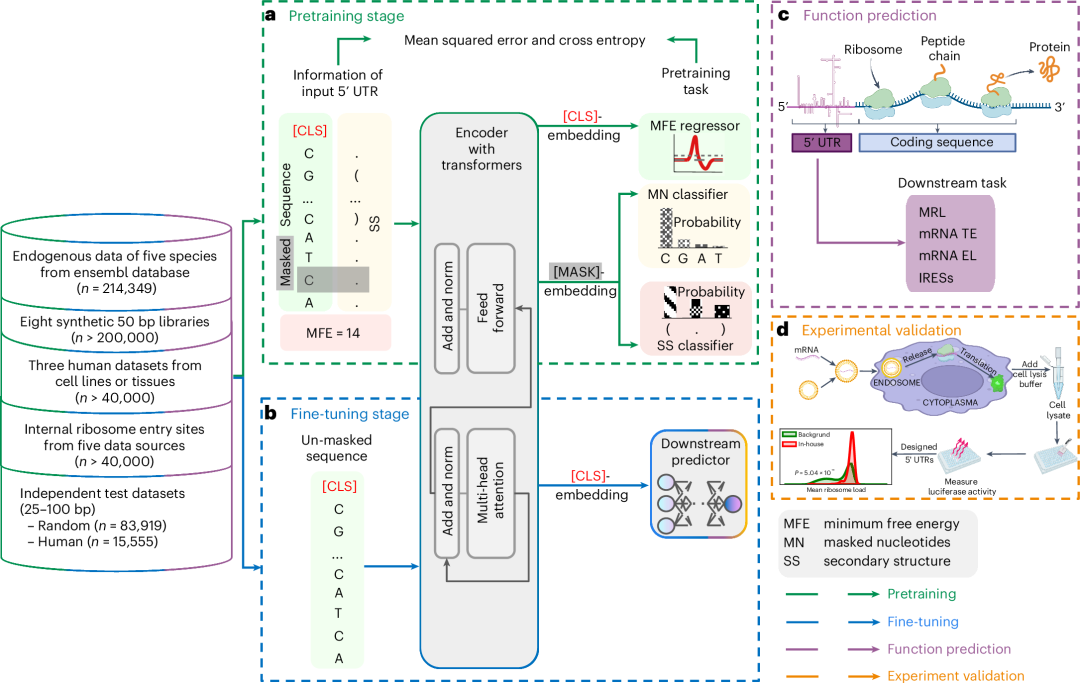
图1. 5′ UTR语言模型(UTR-LM)的开发和应用流程:(a) 模型接收5′ UTR序列作为输入,通过128维随机嵌入和[CLS]-token传输至transformer层,预训练阶段包括掩蔽核苷酸预测(MN regressor)、5′ UTR二级结构预测(SS classifier)和最小自由能预测(MFE classifier);(b) 利用[CLS]-token对模型进行下游任务的训练;(c) 对模型进行微调,以预测平均核糖体负载量(MRL)、mRNA翻译效率(TE)、mRNA表达水平(EL)和内部核糖体入口位点(IRES)等关键指标;(d) 根据模型预测的高翻译效率设计5′ UTR库,并通过mRNA转染和荧光素酶实验进行湿实验室验证。
结果
平均核糖体负载量(MRL)
MRL是指在给定时间内,一个特定mRNA分子上活跃的核糖体数量的平均值。它是衡量mRNA翻译效率的一个重要指标,反映了mRNA被翻译成蛋白质的速度和程度。研究表明,UTR-LM模型在预测MRL方面具有卓越性能,这直接影响蛋白质的生产效率。图2展示了UTR-LM模型与其他基准模型在不同数据集上的性能对比。
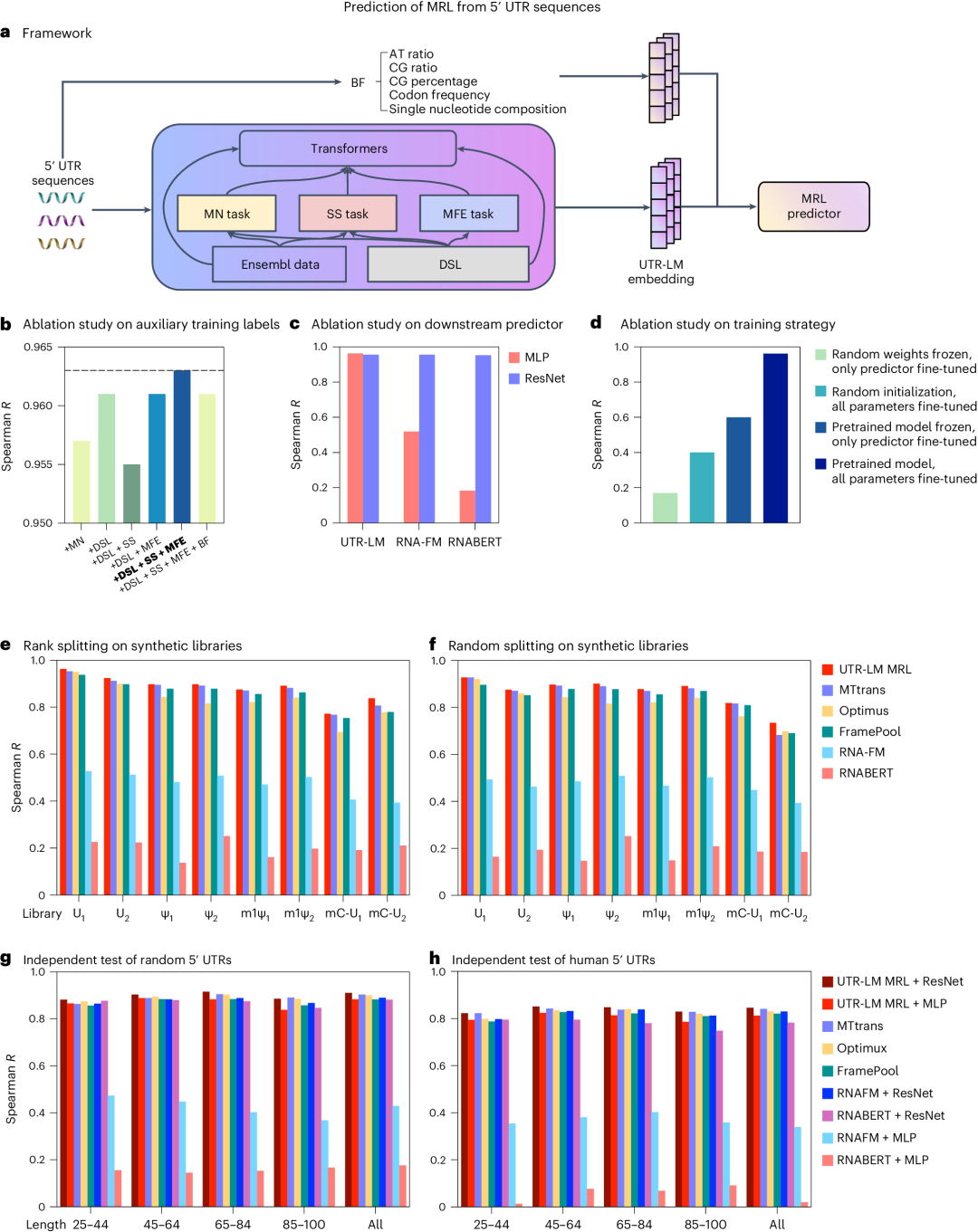
图2. a,UTR-LM 框架,包括在预训练期间集成下游库 (DSL)、二级结构(SS) 和最小自由能(MFE)以及手动提取的生物特征 (BF) 的变体。b,UTR-LM 超参数的消融研究。在后续实验中,使用了由 DSL、SS 和 MFE 增强的基线 UTR-LM,称为 UTR-LM MRL。c,预训练模型在结合简单MLP或复杂下游网络架构(ResNet)后对5′ UTR序列特征提取和功能预测能力的影响。d,评估不同训练策略对模型性能的影响,对比了预训练+微调策略与采用随机权重和/或固定权重的训练策略。e,f,对8个合成的50 bp核苷酸的5' UTR 的文库的各种方法的评估。g,h,使用独立测试评估各种方法。
mRNA表达水平(EL)和翻译效率(TE)
EL通常指mRNA或蛋白质在细胞中的丰度,可以通过各种技术如RNA测序(RNA-seq)来量化,反映了基因转录后产物的累积效果。TE是指mRNA从核糖体翻译成蛋白质的速率,它通常通过比较mRNA分子上的核糖体占用情况(即核糖体足迹)与mRNA在细胞中的丰度来计算。UTR-LM模型准确预测了mRNA的这两项指标,这两个因素对于理解蛋白质生产至关重要。如图3所示,模型在这些任务上优于现有的所有基准方法。
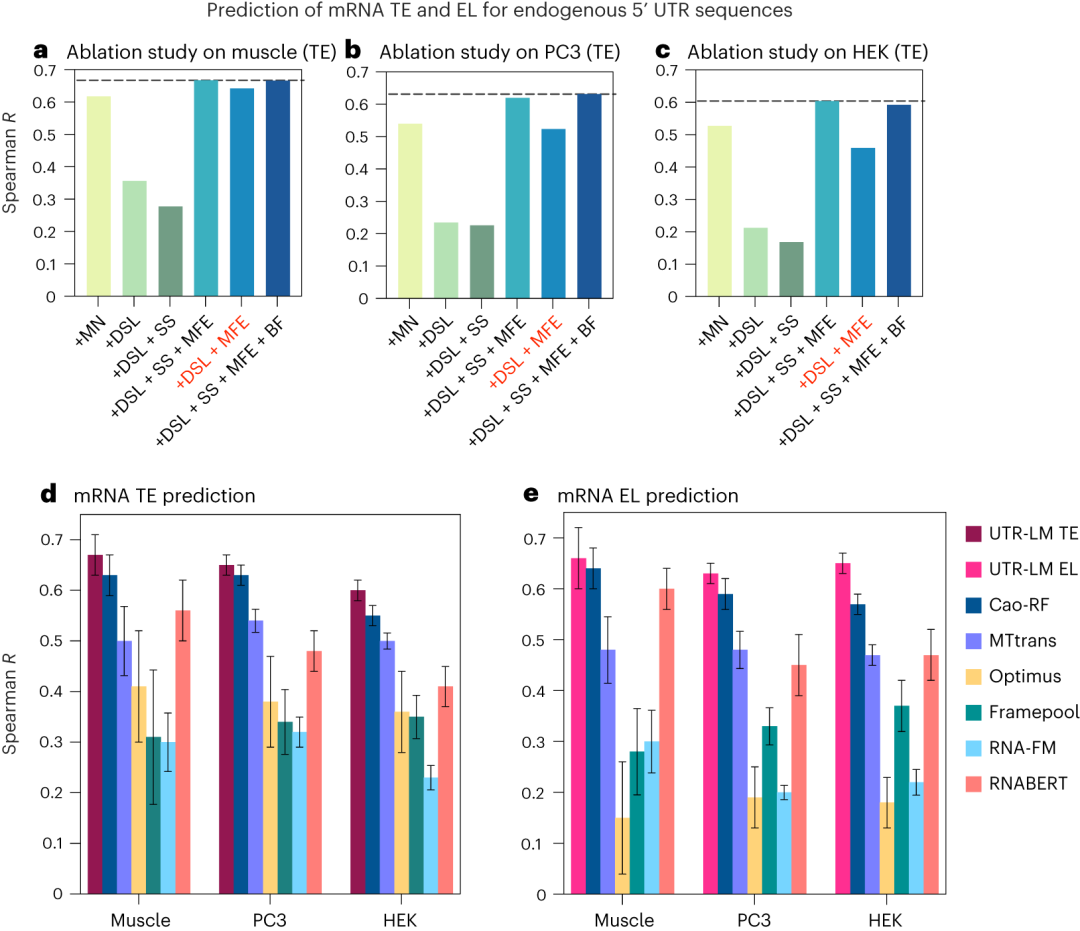
图3. 这些数据集的来源包括人体肌肉组织(Muscle)、PC3 和 HEK293T 细胞系。a-c,UTR-LM超参数在TE任务上的消融研究。在后续实验中,使用DSL和MFE增强的UTR-LM作为最终模型。d,对于 TE 预测,UTR-LM 模型优于 Cao-RF 高达 5%,并且在 Spearman R 方面优于 Optimus 高达 27%。e,对于 EL 预测,UTR-LM 模型优于 Cao-RF高达 8%,并且优于 Optimus 高达 47% 。配对 t 检验表明 UTR-LM 显著优于其他基准(P < 0.05)。
未标注内部核糖体入口位点(IRES)的识别
IRES是mRNA上的一段特殊序列,它允许核糖体直接绑定到mRNA中部并启动翻译,而不是从传统的5′端帽结构开始。IRES存在于某些病毒和细胞mRNA中,它们通过这种机制可以在细胞应激或病毒感染时调控蛋白质的合成,使翻译过程能在帽依赖性翻译受阻的情况下继续进行。通过UTR-LM模型,研究团队成功识别了未标注的IRES,这对于理解mRNA的内部翻译启动机制至关重要。如图4所示,UTR-LM在这一任务上的表现显著超越了最佳基准。
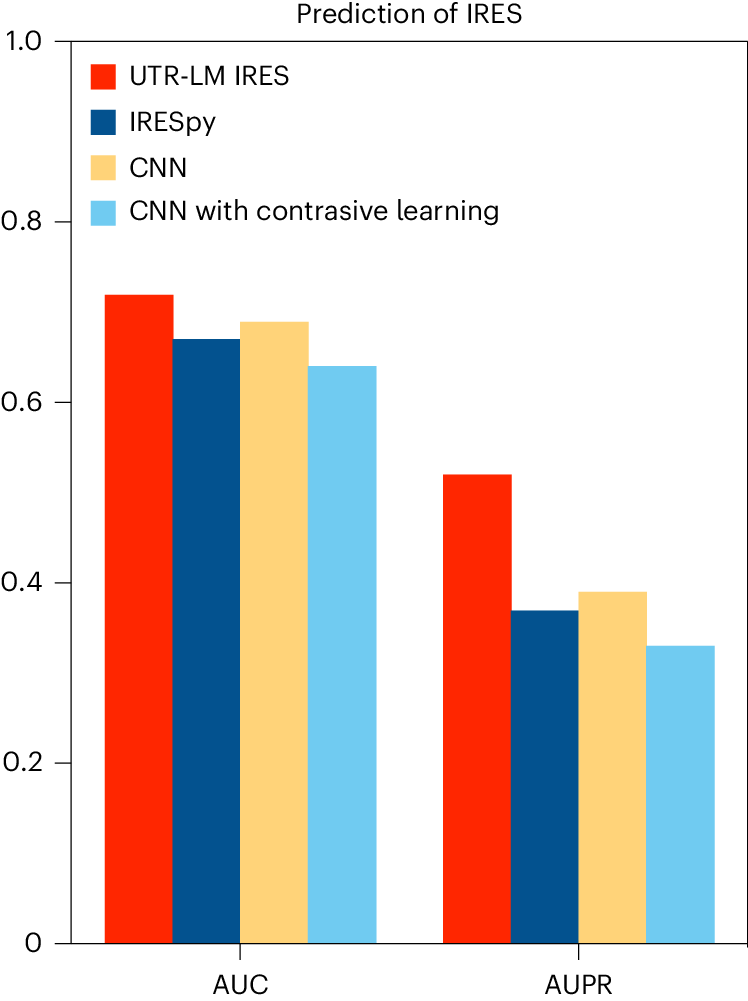
图4. UTR-LM IRES 分类器比该任务的benchmark IRESpy实现了更高的AUPR和AUC。所有模型均通过十折交叉验证在 46,774 个序列上进行训练和测试。
湿实验验证
通过设计并测试新的5′ UTR,研究验证了UTR-LM模型的预测准确性。图5表明,新设计的5′ UTR能够有效提高蛋白质的生产水平,证明了模型在生物技术和治疗领域的应用潜力。
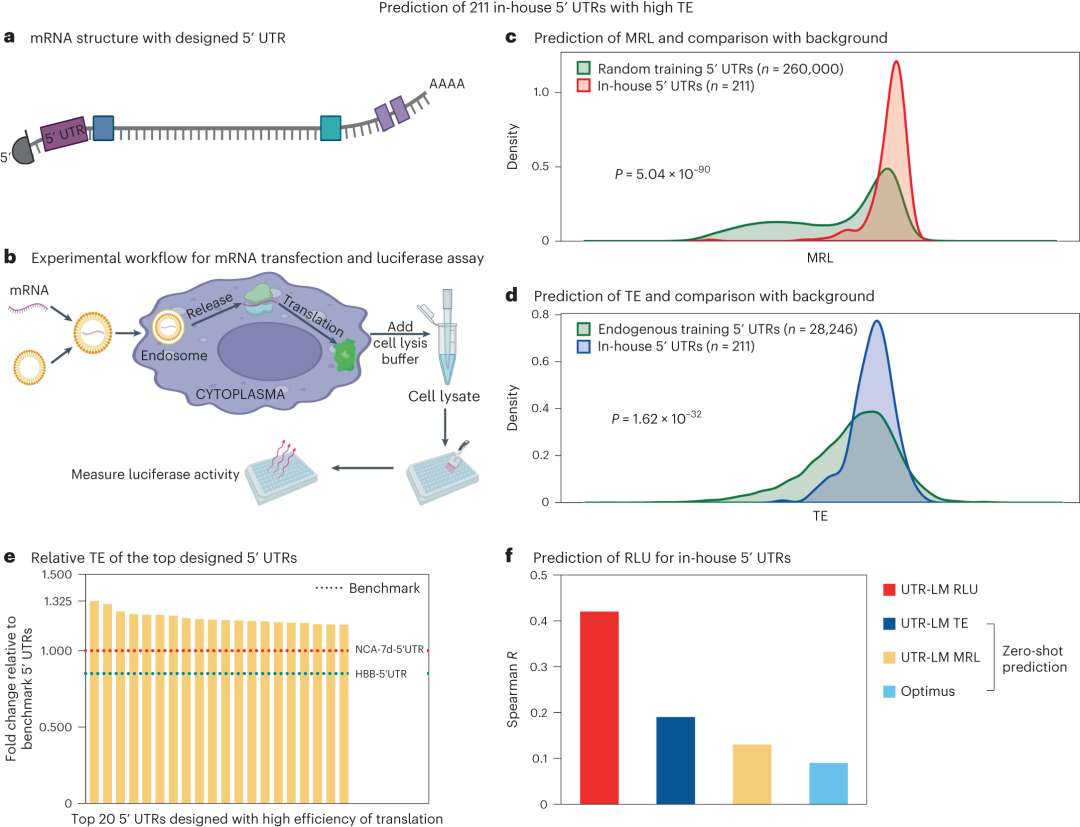
图5. 通过湿实验设计并验证了包含211个具有高TE的5' UTR库。这些序列随后被用作独立的测试集来验证模型的效率。a,内部设计的5' UTR的mRNA结构。b,mRNA 转染和荧光素酶测定的实验流程。c,d,与背景相比,内部5' UTR具有更高的 MRL (c) 和 TE (d) 预测值。e,湿实验结果:前 20 个设计的5' UTR与基准5' UTR NCA-7d-5'UTR和HBB-5'UTR相比的相对TE。f,使用湿实验结果作为独立测试集,与基准相比,UTR-LM 给出了更准确的zero-shot prediction。
模型的生物学可解释性
在这项研究中,通过一系列深入分析, UTR-LM展示了其在识别序列特征、定位功能影响区域以及发现功能性基序方面的强大能力。首先,通过Sequence Logo揭示了五个物种5′ UTR区域的核苷酸组成和分布独特性,高亮了物种间序列水平的差异。轮廓系数(Silhouette Score)的应用表明,UTR-LM模型能够有效区分不同物种的5′ UTR,验证了其在生物多样性研究中的应用潜力。UMAP进一步揭示了模型可以识别出序列的隐藏信息,比如MFE。此外,对单个实例和15,555个人类5′ UTR序列的注意力分数分析揭示了对MRL有重大影响的关键序列区域。成功识别Kozak序列(KCS)及其保守模式CCACC进一步证实了UTR-LM在识别对基因表达和蛋白质翻译具有重要影响的序列元素方面的有效性。
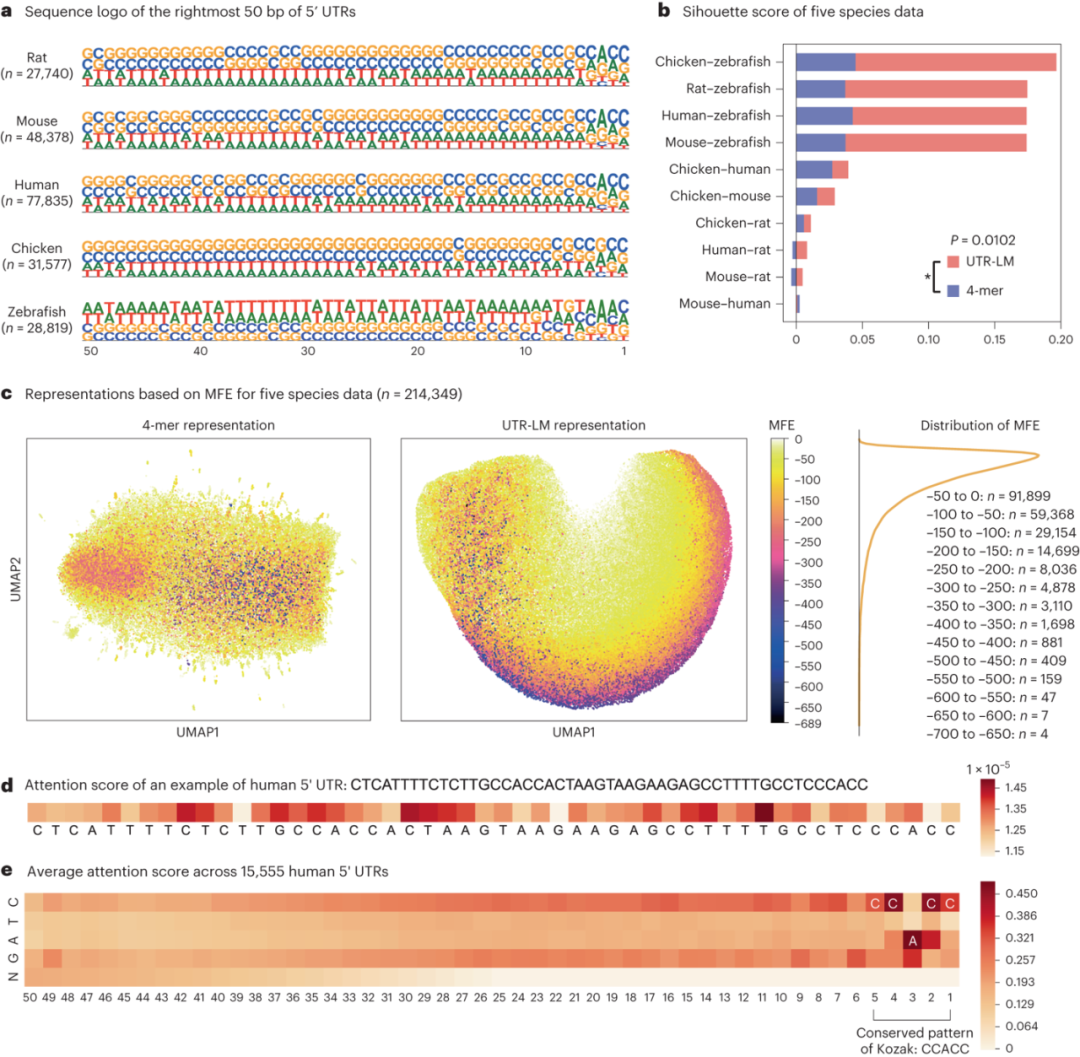
图5. a,五个物种的Sequence Logo。b,UTR-LM更高的Silhouette score表明:相比简单的4-mer表示,语言模型可以对不同物种进行更好的区分。c,由MFE着色的UMAP可视化揭示了UTR-LM可以识别序列的隐藏特征。d,人类5' UTR实例的注意力分数,其中较高的分数意味着相应位点对MRL的预测具有较高的影响。e,15,555 个人类5' UTR中每个位置和核苷酸的平均注意力分数,揭示了KCS及其保守模式CCACC,这是一个在很大程度上影响MRL的已知基序。
结论
这项研究通过开发UTR-LM模型,提高了对mRNA调控区功能解码的准确性,为蛋白质表达的预测和优化提供了新的视角。该模型在核糖体装载、翻译效率和表达水平预测等多个生物学任务上展现出优越性能,并通过湿实验验证了其预测的新5′ UTR设计的有效性。研究不仅拓展了对mRNA 5′ UTR功能的理解,也为未来的基因调控和疗法创新提供了宝贵的工具和知识基础。
参考资料
Chu, Y., Yu, D., Li, Y. et al. A 5′ UTR language model for decoding untranslated regions of mRNA and function predictions. Nat Mach Intell (2024). https://doi.org/10.1038/s42256-024-00823-9
代码
https://github.com/a96123155/UTR-LM
内容中包含的图片若涉及版权问题,请及时与我们联系删除

评论
沙发等你来抢