
今天给大家介绍的是来自浙江大学公共卫生学院附属第二医院医学院团队在BIB 2024上发表的有关多药联合预测药物协同作用的一篇文章,其主要通过相互关注机制实现跨模态的交互。
本文提出了一个,通过卷积生成细胞系嵌入;构建了互注意模块(DCMA,DDMA和CCMA)和自注意模块多模态互注网络框架 SynergyX,药物特征使用ESPF指纹,细胞系特征中的每个基因使用6种不同特征进行表示(DSSA,CSSA)用于进行跨模态交互,特征提取和提供最终的可解释性。

1.介绍
发现有效的抗肿瘤药物组合对于推进癌症治疗至关重要。充分考虑复杂的生物相互作用对于准确预测药物协同作用非常重要。然而,极其有限的先验知识给当前计算方法的发展带来了巨大的挑战。
为了解决这个问题,本文提出了一个多模态的相互关注网络SynergyX,它动态捕获跨模态相互作用,允许对复杂的生物网络和药物相互作用进行建模。采用卷积增强注意结构对该框架中的多组数据进行有效集成。与其sota模型相比,SynergyX在通用测试和盲测试以及跨数据集验证中都表现出卓越的预测准确性;SynergyX的另一个显著优势在于它的多维可解释性。综上所述,SynergyX提供了一个具有启发性和可解释性的框架,有望催化药物协同作用发现的探索,加深我们对合理联合治疗的理解。
2.方法
2.1 药物亚结构编码
对于每种药物,从ChEMBL数据库中获取其SMILES,然后使用RDKit库将其转换为规范的SMILES。我们进一步将药物的SMILES格式转换为可解释的子结构分区指纹图谱(ESPF),使后续注意模块能够捕获可解释的药效团信息。ESPF通过将药物分解成一组离散的、中等大小的亚结构,形成药物的顺序编码。Huang等人确定了大约2700个有价值的子结构,形成了一个子结构词典。最后,将药物表示为大小为165的定长向量,这里与细胞的后续输入特征的尺寸相匹配。如果药物中的子结构数少于165个,则剩余的位置将用零填充。
按照上述步骤,将每种药物表示为one-hot向量,表示药物序列第 个子结构的子结构索引(该索引是指上述约2700个子结构构成的结构词典中每个子结构对应的index)。根据前人的研究方法,我们进一步对这些特征进行了转化。首先,我们使用一个可学习的字典查找矩阵
个子结构的子结构索引(该索引是指上述约2700个子结构构成的结构词典中每个子结构对应的index)。根据前人的研究方法,我们进一步对这些特征进行了转化。首先,我们使用一个可学习的字典查找矩阵 ,其中
,其中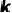 表示子结构的最大数量,
表示子结构的最大数量, 表示向量维数。这样我们就可以将每种药物的子结构索引转换成一个长度为
表示向量维数。这样我们就可以将每种药物的子结构索引转换成一个长度为 的向量(其中
的向量(其中 ),从而得到每种药物的初始特征
),从而得到每种药物的初始特征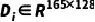 。随后,为了获取药物子结构的位置信息,使用查找字典
。随后,为了获取药物子结构的位置信息,使用查找字典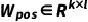 计算位置表示:
计算位置表示: 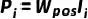
其中, 是一个one-hot向量,第
是一个one-hot向量,第 个位置设为1。因此,我们将初始特征和位置表示相加,生成最终的药物子结构编码:
个位置设为1。因此,我们将初始特征和位置表示相加,生成最终的药物子结构编码: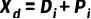
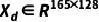 作为后续跨模态编码器的输入。
作为后续跨模态编码器的输入。2.2 统一基因集
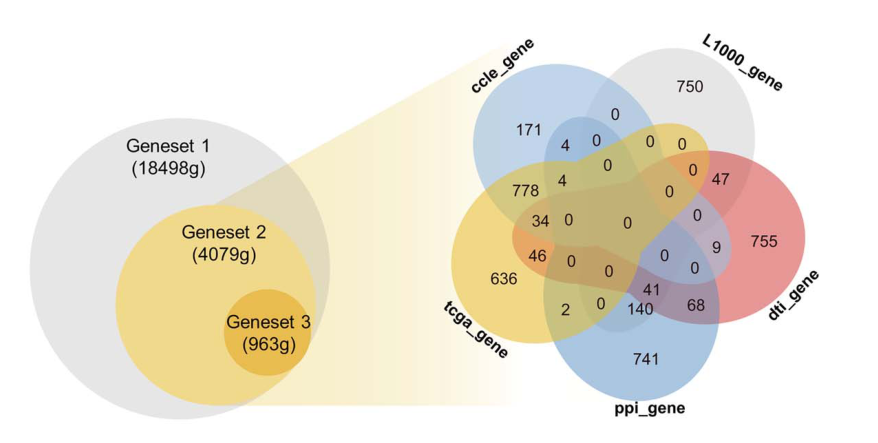
为了确定一个潜在的有助于药物协同作用的重要基因子集,我们从不同的集合中选择标记基因并整合它们。LINCS项目发布了一个包含978个基因的里程碑式基因集,已知该基因集捕获了整个转录组81%的信息。这些基因包含在我们最终的基因集中。此外,我们选择了CCLE项目中癌细胞株基因表达变异最大的前1000个基因和TCGA中肿瘤样本中变异最大的1500个基因。这些基因可以有效地捕捉不同样本的异质性。
此外,基于STRING数据库中包含的PPI网络,我们过滤了综合得分高于0.7的相互作用,然后确定了与其他蛋白质相互作用最多的前1000个蛋白质。编码这些蛋白质的基因通常被认为在生物网络中更为关键和功能重要。此外,从STITCH数据库选择了1000个与最多药物相关的药物靶向基因。最后,我们将上述所选择的5组基因(L1000_gene、ccle_gene、tcga_gene、ppi_gene和dti_gene)进行组合,去掉缺失特征的基因后,得到4079个基因。
2.3 细胞系的多基因组整合
SynergyX利用了从DepMap数据库下载的6种组学数据[39,40]:基因表达(exp)、基因突变(mut)、基因拷贝数(cn)、基因甲基化(met)、基因效应(eff)和基因依赖概率(dep)。基因突变数据被处理为二进制变量,其中0代表正常基因,1代表突变基因。对于每个细胞系,我们的目标是保留上述4079个基因的组学特征。虽然我们收集了所有167个细胞系中exp、mut和cn的完整基因特征,但仍有缺失其余特征数据的情况。其中eff和deep在138个细胞系中收集了3456个基因,met在143个细胞系中收集了2279个基因。如果缺少任何组学特征,则用该基因在其余细胞系中的平均值进行计算。我们应用tanh归一化,类似于DeepSynergy,对不同组型的原始数据进行预处理。对于一个特定的细胞系,6个组学特征按照固定的基因顺序整合,得到一个输入特征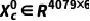 。
。
受卷积增强Transformer结构(Conformer)的启发,我们采用一维卷积神经网络(CNN)在注意模块之前对细胞系的原始多组数据进行初步整合,考虑到细胞系特征序列相对较长,我们在卷积层之间引入了maxpooling的子采样。CNN擅长提取局部特征,当与擅长建模长期全局上下文的级联注意力模块结合时,它们可以更充分地表示细胞系特征。经过CNN模块,我们获得了细胞系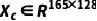 的综合多组学特征。
的综合多组学特征。
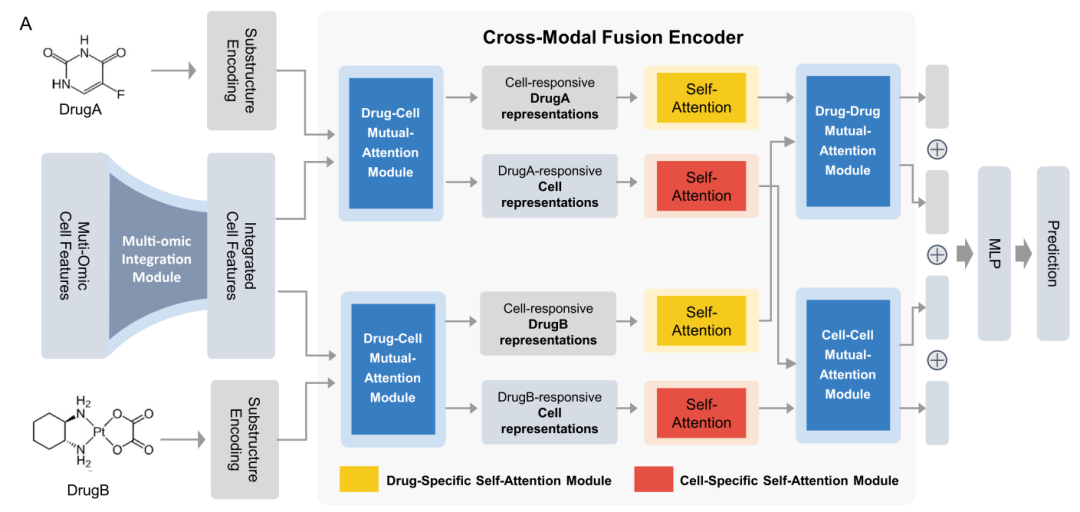
2.4 跨模态融合编码器
Synergy的核心模块是跨模态融合编码器,该模块采用多种注意力模块实现特征更新和融合,同时提取潜在的药物-细胞和药物-药物相互作用。我们尝试了相互关注和自我关注模块的不同组合,并最终在我们的SynergyX中确定了一个三层“三明治”结构。外层是相互关注模块,中间层是自关注模块。
2.4.1 跨模态互注意力
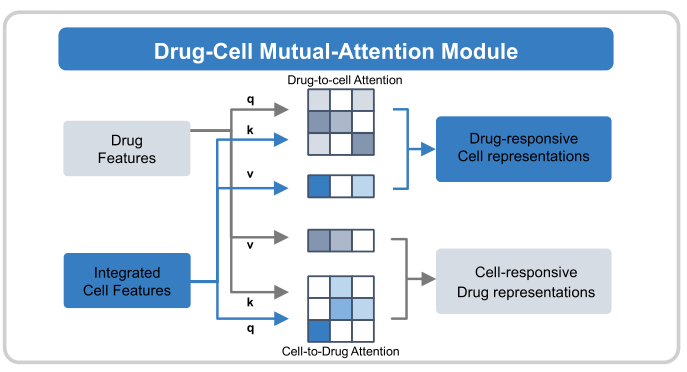
跨模态融合编码器从药物-细胞相互注意(DCMA)模块开始,用于跨模态相关特征提取。如图3所示,DCMA模块由多头互注意网络和前馈神经网络组成。核心步骤包括计算药物与细胞和细胞与药物相互作用的权重。药物-细胞注意矩阵用于获得药物反应性细胞潜伏表征。相反,细胞-药物注意矩阵用于获得细胞反应性药物潜伏表征。
2.4.2 特定特征的自注意力更新
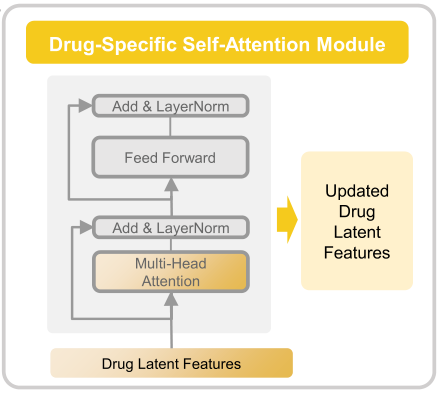
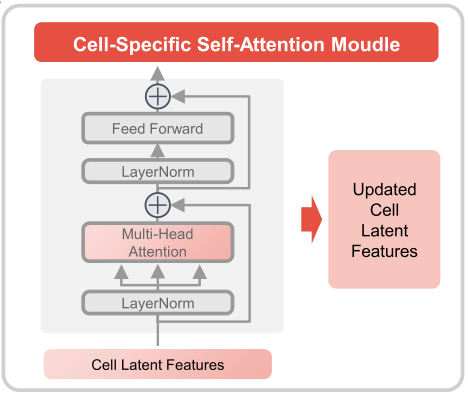
2.4.3 单向互注意力
在第三层,我们使用两个单峰互注意模块来提取粗粒度的融合信息。其中,药物-药物相互关注模块DDMA整合了药物A和药物B的潜在特征。细胞-细胞互注意模块(CCMA)分别用于结合对药物A和药物B有反应的细胞潜伏特征。DDMA和CCMA都由多头互注意层和前馈神经网络组成。
2.5 预测模块
3.实验
3.1 数据集
药物组合数据来自综合数据库DrugComb (v1.5),该数据库包含739964个组合实验。它提供了一种新的药物协同作用测量方法称为S评分,它通过测量药物组合与单一药物的剂量反应曲线之间的差异来定义药物协同作用的水平。研究表明,与现有的四种协同作用评分(HSA、Bliss、Loewe和ZIP)相比,S评分在预测最具协同作用和拮抗作用的药物组合方面具有较高的准确性(AUC>0.99)。我们选择S评分作为药物对在特定细胞系中相互作用的定量指标。
我们从DrugComb数据库中选择了所有可用的数据进行进一步的数据清理。首先,我们删除了关于药物和细胞系信息不清楚或缺失的条目。接下来,我们在数据集中发现了一个明显的数据不平衡问题,其中2157种药物(85.9%)出现的次数少于10次,仅占数据集的1.37%(4587项)。为了解决这种数据不平衡并提高数据集的质量,我们消除了出现次数少于10次的药物。此外,我们采用3σ原理来识别数据集中的异常值,然后使用平均值±3个标准差作为阈值,并为该范围之外的分数分配边界值。最终,我们获得了330917种药物组合的数据集,涉及354种药物和167个细胞系。值得注意的是,尽管DrugComb是现有最大的药物组合数据库之一,但与预测空间相比,标记的数据仍然显着较小。在我们的案例中,167个细胞系中354种药物的组合空间相当于大约2100万种可能性,而现有数据仅覆盖了总可能性的1.56%。
3.2 模型评估
为了评估SynergyX的性能,我们将其与六种具有代表性的最先进的深度学习方法和两种杰出的机器学习方法Random Forest(RF)和XGBoost进行了比较。所有方法都在用于SynergyX的相同数据集上进行了训练和评估。提到的六种深度学习方法分别是DeepSynergy、MatchMaker、DeepDDS、DTSyn、MGAE-DC和DFFNDDS。对于每种方法,我们都试图保持各自研究中提到的一致的输入特征、模型架构和最优训练参数。具体来说,DeepDDS使用GCN或GAT进行药物特征提取,我们将这两种模型分别命名为DeepDDS-GCN和DeepDDS-GAT。
此外,对于最初为分类任务设计的模型,如DeepDDS和DFFNDDS,我们对它们的预测模块和损失函数进行了轻微的调整。我们还在合理的范围内优化了它们的训练参数,使其更适合于回归任务。针对RF和XGBoost,采用类似网格搜索的方法寻找最优训练参数。值得注意的是,cuML包被用来利用GPU加速来训练RF模型
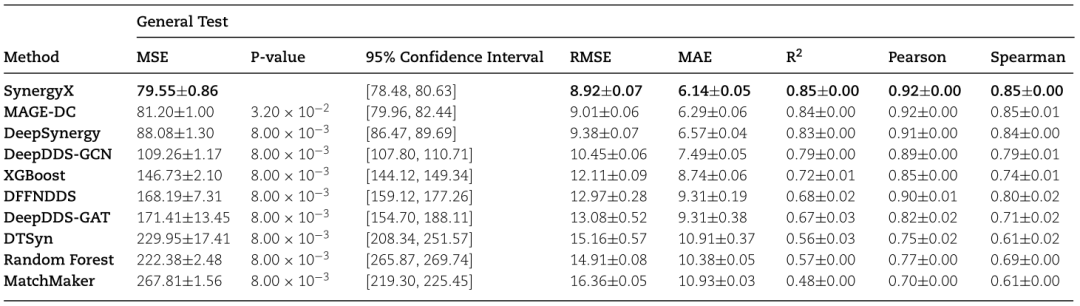
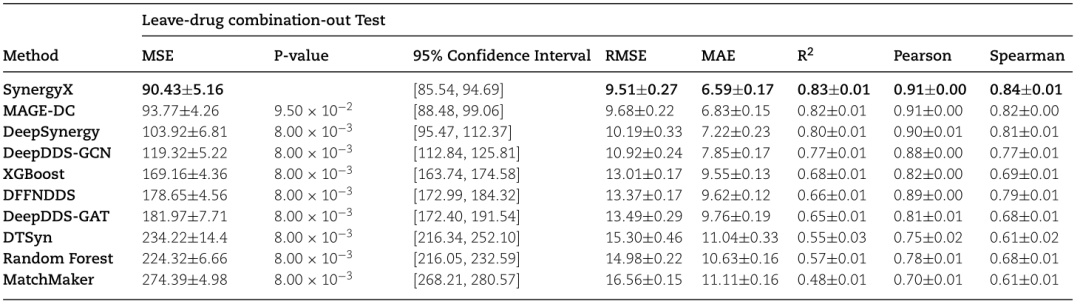
在两种实验设置中对SynergyX和所有比较方法进行评估:(1)重新发现已知药物协同作用的一般测试,(2)发现未知药物对的遗漏药物组合测试。在General测试中,将整个数据集按8:1:1的比例划分为训练集、验证集和测试集。对于遗漏药物组合测试,我们采用了基于药物对的分层抽样方法,确保测试集不包括训练集中存在的任何药物对。最终的训练集、验证集和测试集分别约占所有药物对的80%、10%和10%。所有实验用不同的随机种子重复5次。我们使用均方误差(MSE)、均方根误差(RMSE)、平均绝对误差(MAE)、r平方(R2)和Spearman相关性(Spearman)作为回归预测任务的评价指标。此外,我们在四个独立的数据集上进一步评估了SynergyX,以证明其在不同数据集上的强大预测能力。数据集分割策略和实验设置与上述一致。性能比较如表1,2所示:
3.3 消融实验
SynergyX包含6个子模块:CNN (多组学集成模块)、DCMA、DSSA、CSSA、DDMA和CCMA。移除SynergyX的不同组件,以评估它们对整体性能的贡献。变体模型被标记为SynergyX-B、SynergyX-C、SynergyX-D、SynergyX-E、SynergyX-F和SynergyX-G。每个模型进行了五次随机实验,以研究某些模块的缺失是否会显著影响模型的性能。如表3所示,完整的SynergyX模型展示了整体最佳性能,这表明了每个组件对模型的贡献的重要性。
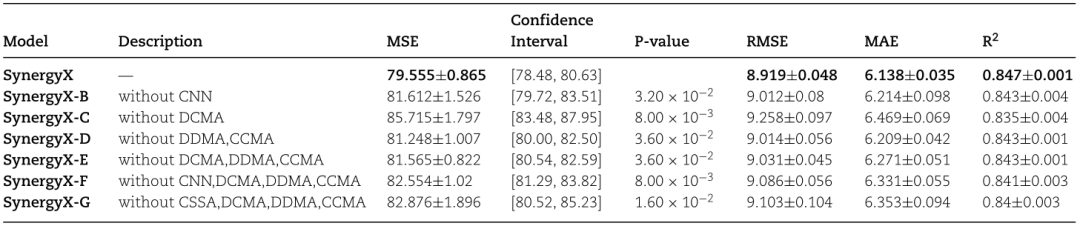
考虑到SynergyX、SynergyX- B、SynergyX-E、SynergyX-F和SynergyX-G,我们可以观察到,无论是否存在相互关注模块,卷积增强的注意力架构将CNN和注意力结合起来往往比单独使用其中任何一个都能产生更好的结果。这与我们的假设一致,即CNN特征捕获局部感知特征,而注意力机制可以有效捕获远程语义信息。结合两者可以获得更好的细胞系表示
3.4 多组数据研究
接下来我们想要弄清楚的两个问题是:
(1)使用多组数据是否总是比使用单一组数据更有效?
(2)特定类型的组学数据是否更适合药物协同作用预测?
因此,我们探索了不同组合组学数据的影响。一共有六种类型的基因组最初应用于SynergyX。为了简化实验,我们随机选取了涉及2、3、4和5种基因组数据的6种组合。如图6所示。
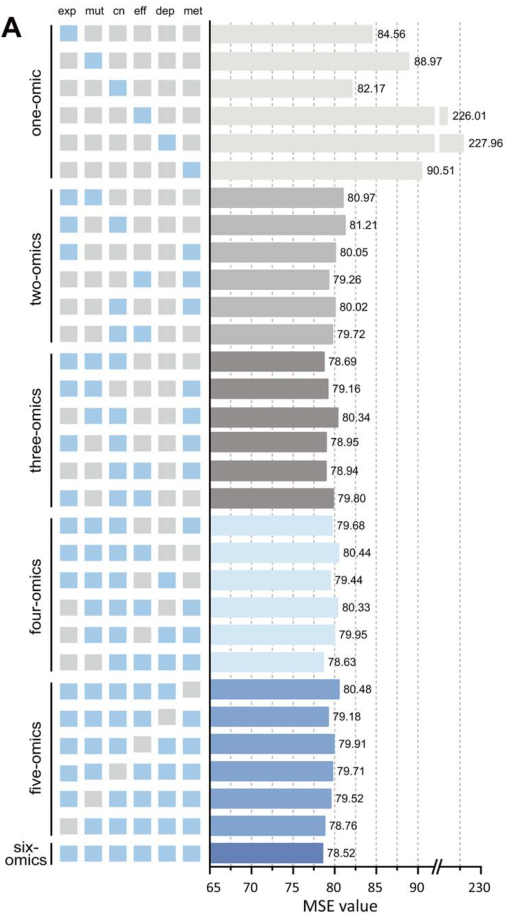
在单组学实验中,不同组学类型的模型性能不同。cn数据的MSE值最低,为82.17,而exp、mut和met表现相对较好。然而,使用eff或dep会导致明显较差的结果。这是因为我们缺乏足够的eff和dep特征,这些特征可以作为补充特征,但在单组实验中应该是无效的。此外,我们观察到,当只使用exp、mut、cn或met数据时,SynergyX始终优于其他模型,这强调了SynergyX架构的优越性,随着组数据类型的增加,模型的预测能力得到一定程度的提高,但当超过两种类型时,SynergyX对不同组合并不敏感,一种可能的解释是,不同的基因组类型提供的信息是互补的,而不是独立的。当所有6个基因组数据都被使用时,获得最佳结果
结论
研究提出一种多模态互注意力网络(SynergyX)用于药物协同作用的预测,性能上看表现优异同时提出了合理的可解释性。SynergyX能够破译药物相互作用的复杂图景,识别环境特异性反应,使更有效地发现药物协同作用。
内容中包含的图片若涉及版权问题,请及时与我们联系删除

评论
沙发等你来抢