
用语言模型进行蛋白结构预测
ESM2 & ESM-fold
作为ESM系列的最后一篇文章,这篇于2023年发表在Science上的文章,提出了ESM-2和ESM-fold模型,向我们展示了蛋白质语言模型是如何一步步优化,并且在蛋白质结构预测问题上发挥重大作用的。机器学习在蛋白质结构预测领域的最新进展,是利用多序列比对MSA中的共进化信息来预测蛋白质结构,如Alphafold2等。而当蛋白质序列的语言模型扩展到150亿参数时,学习到的表征中就蕴含蛋白的共进化信息。使用语言模型的表征能使蛋白结构预测的速度加快一个数量级,从而实现元基因组蛋白质的大规模结构表征。作者利用这种能力构建了ESM元基因组图谱,预测了超过6.17亿个元基因组蛋白质序列的结构,其中包括超过2.25亿个高置信度预测序列,从而让人们看到了天然蛋白质的丰富多样性。
编辑|SHO
论文速览
此论文是ESM-2和ESM-fold两个模型的集成; ESM-fold使得蛋白结构预测的速度提升一个数量级; ESM-fold相比于AF2更适用于无足够MSA信息的蛋白结构预测,例如:从头设计全新蛋白、元基因组蛋白、孤儿蛋白等。
算法原理
ESM2
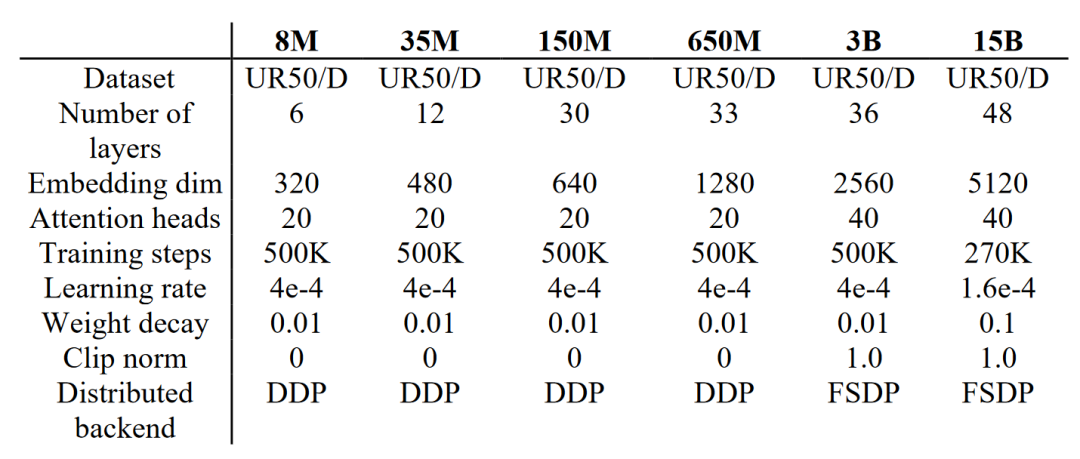
数据集:UniRef50,超6000万个蛋白质序列。
参数量:有不同版本的:8M、35M、150M、650M、3B、15B,参数量越大,perplexity越小,表现越好,15B参数量的perplexity达到了6.37。使用ESM-2预训练时可按需选择不同参数量的模型。
方法:类似ESM-1b,使用了一个Masked language modeling方法,采用了BERT。比较特殊的是在位置编码处作者采用的是旋转位置嵌入(RoPE, rotary position embedding, 提高大模型的外推性,保证在训练和预测时输入的序列长度不同时,模型泛化和鲁棒性不受影响。略微增加计算成本,但是有效。
问题描述:对于15%的随机屏蔽的氨基酸进行预测。
ESM-fold
数据集:蛋白结构数据集分为真实结构和预测结构。真实结构从PDB数据库中收集,包括了32万多条分辨率<9埃米的蛋白质链。预测结构通过AF2生成,为了保证结构的可靠性,只取平均pLDDT>70的序列。
方法:包含两个结构,一个是折叠模块(folding module),将上述语言模型ESM-2(3B参数量版本)输出的特征生成representation,因为是一维的特征所以不需要像AF2的evoformer一样使用轴向注意力机制;另一个是结构模块(structure module),一个等变Transformer输入上述模块输出,并输出3D原子坐标。最终预测LDDT、TM、distogram三个信息。
问题描述:如何把ESM-2的输出映射到蛋白质每个原子的3维坐标。
正文内容
进化过程中的蛋白质序列蕴含大量结构和功能信息,作者认为,在整个进化过程中,填补蛋白质序列中缺失氨基酸的任务需要一个语言模型来理解序列模式的底层结构。随着语言模型的表征能力和训练中所见蛋白质序列多样性的增加,有关蛋白质序列生物学特性的深层信息将会出现。作者训练了一个语言模型ESM2,可以直接从序列中快速实现原子分辨率级别的结构预测。这就替代了目前AF2结构预测过程中成本高昂的部分,不再需要多序列比对MSA,并且大大简化了推理的神经网络架构。这样,仅推理前向传递的速度就提高了60倍,同时还完全省去了搜索相关蛋白序列的过程,而这一过程原先可能需要10分钟以上。
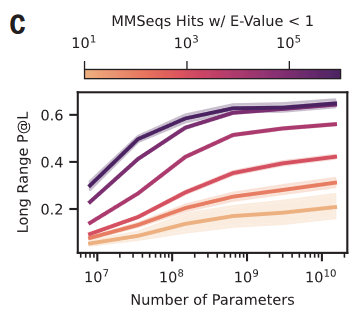
ESM-2参数量的不同对结果的影响可以从perplexity、TM-score和长距离接触准确率预测看出。ESM-2(150亿参数量)在CAMEO测试集上的TMm-score为0.72,在CASP14测试集上的TM-score为0.55,均比1.5亿参数量的中间体提升了15%左右。
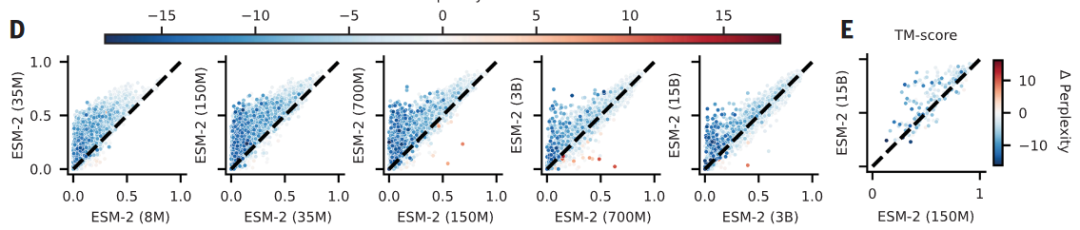
图1,从左到右显示了ESM-2从800万到150亿个参数的模型,通过无监督接触精度作为衡量标准进行比较。
对于相同MMseqs hits,ESM-2(参数量1.5亿)和ESM-1b(参数量6.5亿)的表现相当,说明ESM-2相较于ESM-1b的优越性。
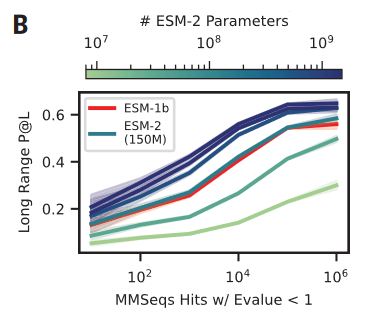
训练集中相关序列越多的蛋白质,其学习轨迹与模型规模的关系越陡峭(图3)。因此,在较低的模型规模下,高进化深度序列(深紫色)的改进会达到饱和,而低进化深度序列(橙色)的改进则会随着模型规模的扩大而持续提高。
基于此,作者进一步开发了端到端的单序列结构预测器ESMFold。预测时,蛋白质序列被输入到 ESM-2。序列通过语言模型的前馈层进行处理,然后将模型的表征传递给折叠头。折叠头从一系列折叠块开始。每个折叠块交替更新序列表征和成对表征。这些块的输出被传递给等变结构模块,并在输出最终的原子级结构和预测置信度(图4A)。
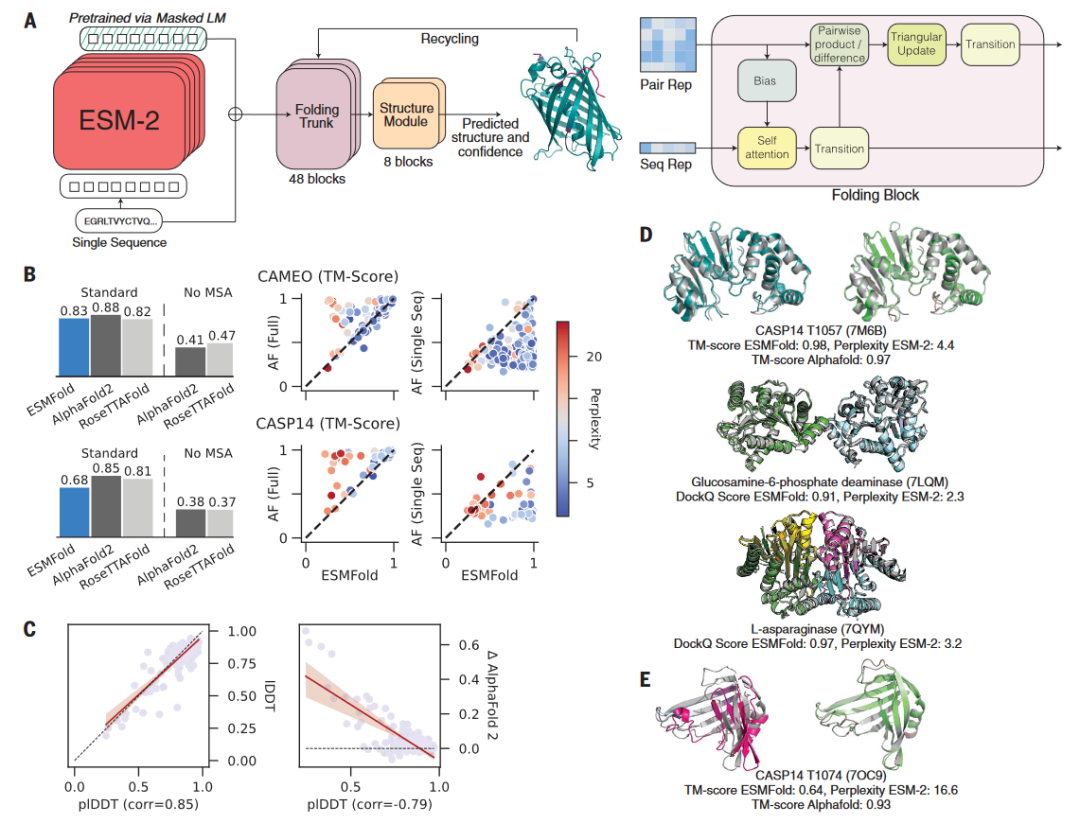
4. 蛋白无MSA时,ESMfold预测准
作者在CAMEO和CASP14测试集上对ESMfold进行测试,平均TM-score分别为0.83和0.68;AF2(包括MSA和模板)的分数为0.88和0.85,但是当删除MSA后再使用AF2进行预测,这一分数分别掉到了0.41和0.38。对于从头蛋白设计而言,大多的蛋白都是自然界中没有存在的,更没有MSA,因此ESMfold将比AF2更适用于此类不存在足够MSA的蛋白(从头设计蛋白和孤儿蛋白)的结构预测。(图4B、C)。
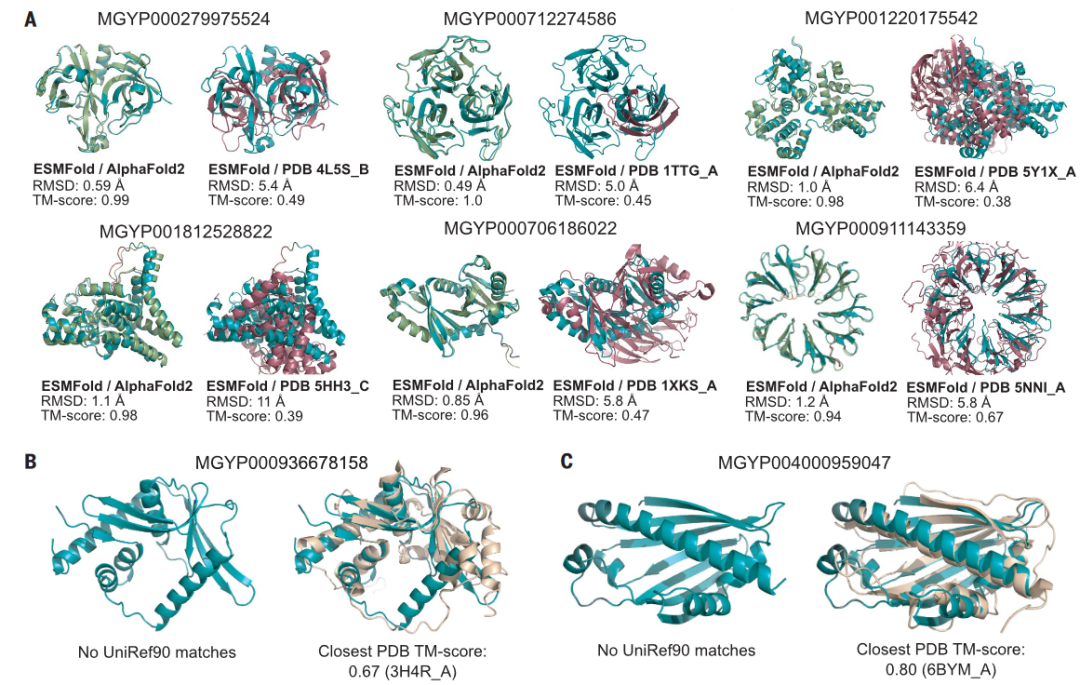
图4,元基因组序列的 ESMFold 结构预测示例。(A) 六种不同元基因组序列的结构预测示例(B和C)两个ESMFold预测结构的例子,这两个结构与PDB中的实验结构有很好的一致性,但与UniRef90中的任何序列的序列同一性都很低。(B)MGYP000936678158 的预测结构与细菌核酸酶的实验结构一致(浅棕色,PDB:3H4R),而(C)MGYP004000959047 的预测结构与细菌固醇结合结构域的实验结构一致(浅棕色,PDB:6BYM)
作者用两周时间在2000个GPU的集群上对MGnify90的数据库中超6.17亿条蛋白进行结构预测,有1.13亿个达到了极高置信度阈值。这些蛋白在Uniref90中已有序列相似度相当低,与PDB数据库中的已有结构相似度也相当低。
论文总结
此论文是ESM-2和ESM-fold两个模型的集成; ESM-2利用语言模型的优势,通过大量的蛋白质序列信息学习蛋白质的embedding表示; ESMfold接着ESM-2学习得到的特征信息投影,生成蛋白质中的原子3D坐标。 蛋白质语言模型训练出的注意力图谱与残基-残基接触图谱有对应关系。作者通过一个线性映射就把语言模型注意力图和结构预测模型的残基残基相互作用图联系起来,这是一个相当有趣的发现。而通过进一步的ESMfold的训练,这一接触图的精度达到了原子级。 ESM-2的作用相当于是替代了AF2和Rosettafold中所需的MSA,学习了一套蛋白质进化与序列-结构规律的通用规则。ESM-fold相比于AF2更适用于无足够MSA信息的蛋白结构预测,例如:从头设计全新蛋白、元基因组蛋白、孤儿蛋白等。 ESMfold相比于AF2的优点: 第一:快,计算速度快约6-60倍不等 第二:不需要搜索目的序列的MSA,进一步节省了大量的时间。
代码文献
Lin, Zeming, et al. "Evolutionary-scale prediction of atomic-level protein structure with a language model." Science 379.6637 (2023): 1123-1130.
https://github.com/facebookresearch/esm
往期文章
关注我们
死磕自己,愉悦大家
专注于AI蛋白相关的论文解读&学术速运
内容中包含的图片若涉及版权问题,请及时与我们联系删除


评论
沙发等你来抢