

蛋白激酶是细胞信号传导途径中的关键调节组件,其异常激活或过表达与多种疾病相关,例如癌症、糖尿病、免疫失调和神经系统疾病等。因此,激酶靶向抑制剂是一类极具价值的治疗药物。然而,由于疾病通路的代偿机制和激酶家族结构相似性带来的脱靶效应,针对单一激酶的抑制剂往往难以持续发挥疗效并带来非预期的毒副作用,而多激酶选择性抑制剂能够在最小化多重药理学风险的同时提供更好的疗效。为了满足更广泛的患者治疗需求,降低激酶脱靶毒副作用风险,研究人员需要对激酶抑制剂的多重药理学效应进行系统评估。目前,激酶组范围的实验活性分析成本高昂,现有的计算工具对未充分研究的“暗”激酶及激酶突变体缺乏预测能力。因此,需要开发具有更广泛应用场景的AI算法,通过充分利用内部或自有实验数据,大规模筛选潜在的多靶点选择性激酶抑制剂。
中国科学院上海药物研究所郑明月课题组建设了一个支持用户定制的激酶多靶点活性筛选AI平台KinomeMETA(https://kinomemeta.alphama.com.cn/,图1)。该平台使用结合图神经网络的元学习技术,极大地拓展了可准确预测的激酶范围,并为用户提供了使用私有数据定制模型的实用功能。研究结果于5月16日在线发表于Nucleic Acids Research,题为“KinomeMETA: a web platform for kinome-wide polypharmacology profiling with meta-learning”。

1.1 高精度、大范围的激酶抑制预测功能
KinomeMETA的预测(Prediction)模块可以预测用户提交分子对661个野生和突变激酶的抑制活性概率。与研究团队先前的小分子激酶多重药理学预测平台KinomeX(https://kinome.simm.ac.cn/en/)[1]相比,KinomeMETA的训练数据和激酶预测范围得到了显著扩展——生物活性数据点增加了3.6倍,化合物增加了5倍,激酶增加了1.69倍。激酶可预测范围的增加得益于元学习算法,该算法使得KinomeMETA能够高效地利用稀疏活性数据对研究较少的激酶和突变激酶提供准确预测。
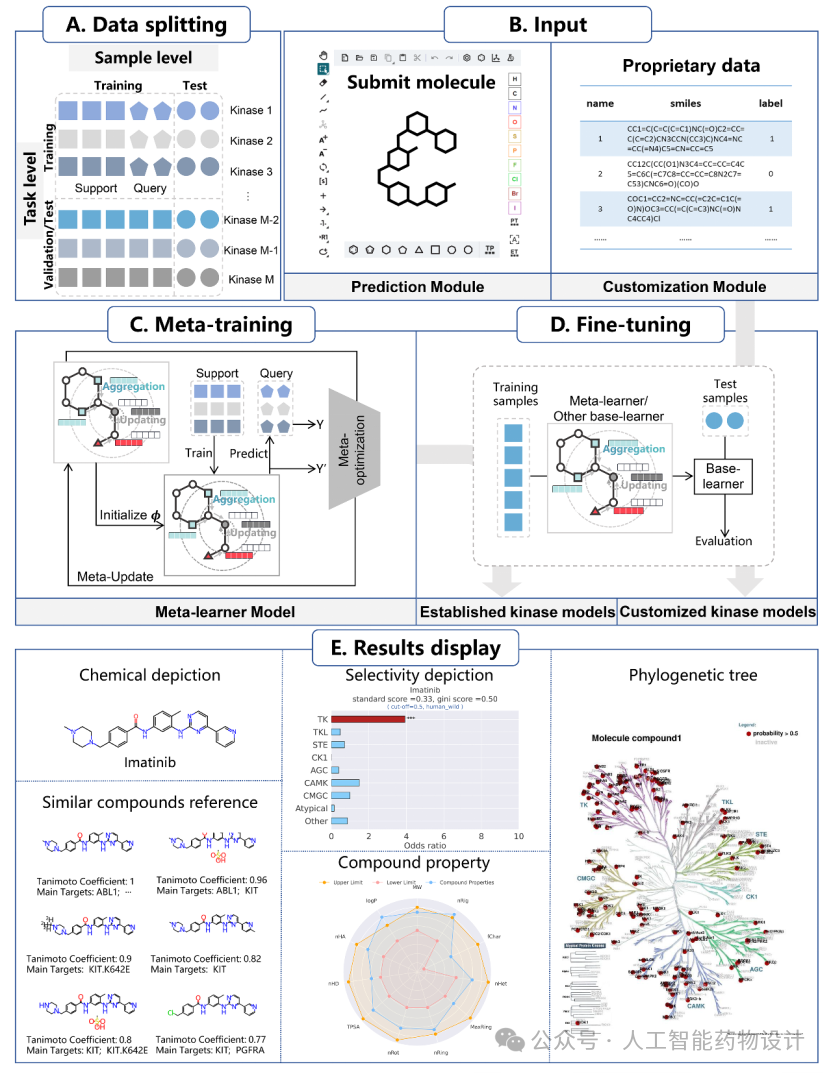
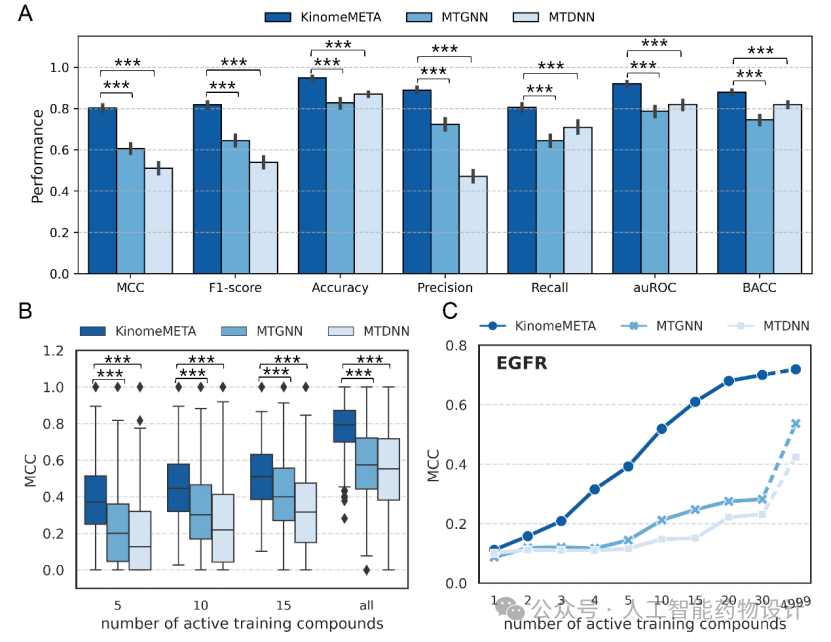
研究人员采用了两种基准方法与KinomeMETA平台进行了性能对比:一种为KinomeX平台使用的多任务深度神经网络(MTDNN),另一种是当前的最先进方法,多任务图神经网络(MTGNN)[2]。对比模型遵循与KinomeMETA相似的构建和训练流程。所有模型在519个测试激酶任务上进行了独立测试。结果表明,KinomeMETA的分类能力始终显著优于MTDNN和MTGNN(图2A)。
用户定制(Customization)模块是KinomeMETA平台的全新功能。该功能允许用户使用少量私有激酶活性数据在线建立定制模型(图1B,D)。这使得KinomeMETA摆脱了传统模型中仅能依靠公开实验数据构建的限制,帮助用户充分结合计算模型与已有实验数据,提升针对特定激酶的研究效能。
在低数据可用性的场景下,传统计算方法通常难以获得对新激酶的泛化能力。与之相比,基于元学习的KinomeMETA通过在多个代表性激酶任务上训练,得到一个具有最优初始化参数的元学习器。元学习器具备对不同激酶的快速泛化能力,仅使用少量数据对其微调即可迅速获得对相应激酶的预测性能[3]。
图2B显示的数据添加实验对比了不同模型在使用不同数量训练数据时的预测性能差异,展示了KinomeMETA利用有限数据快速微调的强大特性。具体而言,研究人员选择了至少有20个活性数据点的测试激酶任务,在每个任务中固定20%的“测试样本”,并分别使用不同数量的剩余活性数据微调模型。,无论所用微调数据量的大小,KinomeMETA的表现始终优于基准模型。以EGFR任务为例(图2C),即使只利用20个活性数据点,KinomeMETA也取得了可观的性能(MCC = 0.68),接近使用全量数据时的性能(MCC = 0.72)。相比之下,基准模型性能提升缓慢且最终性能较低。
1.3 激酶选择性和分子性质分析
除了激酶抑制预测活性外,KinomeMETA平台提供了三个维度的结果分析:激酶选择性、分子性质分析和相似结构抑制剂鉴别(图1E)。
KinomeMETA平台使用标准分数[4]和基尼系数[5]评估提交分子的整体激酶选择性,使用比值比(odds ratio,OR)衡量其对激酶亚家族的选择性[6]。
分子性质分析用于考察提交化合物的类药性,包括提交小分子的预测物理化学性质和药物化学规则评估。我们还开发了一种判别器,用于判断提交化合物是否属于激酶抑制剂。
此外,结果界面还展示数据库中与提交分子结构相似度排名前10的激酶抑制剂及其主要靶点信息,以帮助研究人员探索提交化合物的潜在靶点。
2 案例研究
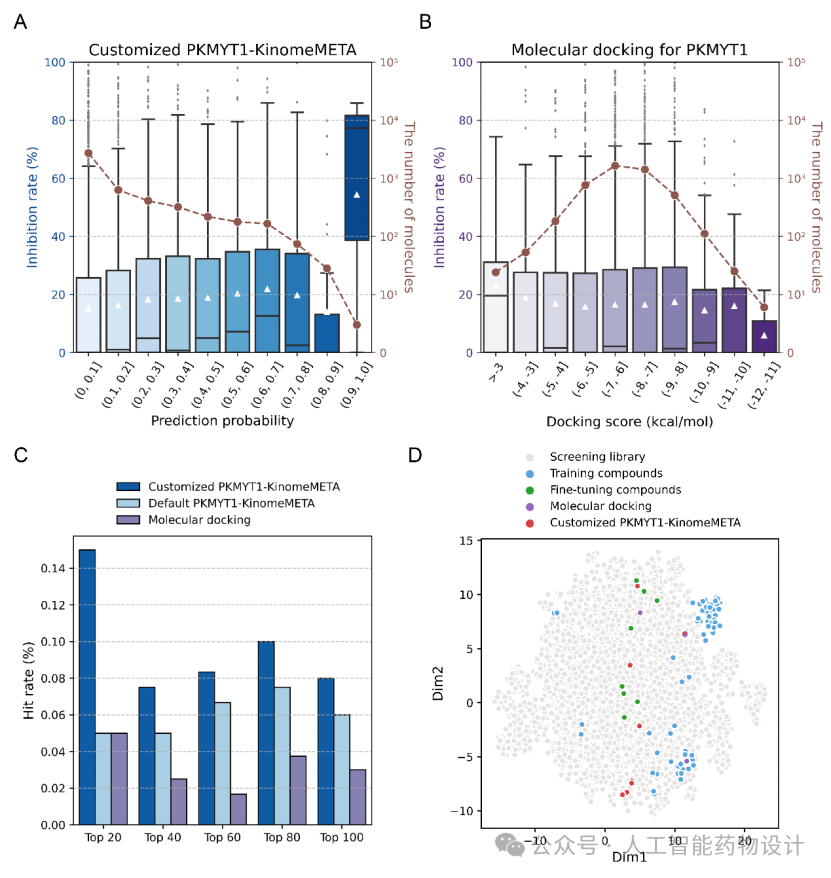
PKMYT1是一个具有高度选择性的激酶治疗靶点,在已有的实验筛选中命中率很低(4.17% [7]和1.25% [8])。在先前的研究中,我们利用KinomeMETA发现了9个新的抑制PKMYT1的活性化合物[3]。
为了说明KinomeMETA定制功能的快速微调特性和实用性,这些化合物被用于微调默认模型(Default PKMYT1-KinomeMETA)以得到新的定制模型(Customized PKMYT1-KinomeMETA)。然后,我们基于KinomeMETA和分子对接两种手段,分别对实验室内部的4,700个小分子化合物进行对PKMYT1的虚拟筛选,并通过实验测定所有化合物在25μM浓度下对PKMYT1的抑制率,以检验两种方法对新活性分子的发现能力。
图3A展示了定制模型预测概率与实验抑制率之间的关系。其中,预测概率超过0.9的化合物显示出更高的抑制率。相比之下,对接模型没能显示出“对接得分更好则命中率更高”的趋势(图3B)。进一步的命中率分析显示,KinomeMETA模型(包括默认和定制)对排名前20至前100的不同范围化合物的命中率均明显优于对接模型(图3C)。此外,定制模型始终优于默认模型,突显了KinomeMETA使用有限新数据快速增强预测性能的高效能力。
基于UMAP对分子进行化学空间可视化分析表明(图3D),通过定制模型新发现的活性化合物数量较多且分布广泛。相比之下,分子对接只发现了3个活性化合物,其中一个与已知活性化合物结构相似,另一个也可被定制模型预测得到。
3 总结
KinomeMETA是一个全面升级的激酶多靶点小分子抑制剂预测平台。相比于先前的KinomeX平台,KinomeMETA拥有更广泛的激酶预测范围和更高的预测精度。本平台的一个重要突破是引入了元学习驱动的定制模块。该模块使研究人员能够使用少量私有数据在线创建新激酶模型或改进现有激酶模型,通过迭代的干/湿实验循环,有效应对药物发现中的低数据场景。KinomeMETA平台将成为激酶研究人员的强力工具,显著加速新型激酶抑制剂的发现。
德州学院李召军博士、中国科学院上海药物研究所研究生曲宁和上海科技大学研究生周敬怡为论文的共同第一作者。中国科学院上海药物研究所药物发现与设计中心(DDDC)李叙潼副研究员、郑明月研究员与张素林副研究员为论文通讯作者。本研究得到了国家自然科学基金、国家重点研发专项、中国博士后科学基金、中国科学院青年创新促进会、上海药物研究所与上海中医药大学中医药创新团队联合研究项目、以及上海市科技重大专项资助。
参考文献
Li. et al., KinomeMETA: a web platform for kinome-wide polypharmacology profiling with meta-learning, Nucleic Acids Research (2024), https://doi.org/10.1093/nar/gkae380.

内容中包含的图片若涉及版权问题,请及时与我们联系删除

评论
沙发等你来抢