
今天为大家介绍的是来自Attila Gursoy团队的一篇论文。蛋白质是生物世界的重要组成部分,具有多种功能。它们通过界面与其他分子相互作用,并参与至关重要的细胞过程。这些反应的破坏可能对生物体产生负面影响,这突显了研究蛋白质-蛋白质界面以开发针对性治疗方法的重要性。因此,开发一种可靠的方法来研究蛋白质-蛋白质相互作用至关重要。在这项工作中,作者提出了一种利用学习到的界面表示来验证蛋白质-蛋白质界面的方法。该方法涉及使用基于图的对比autoencoder架构和transformer来从无标签的数据中学习蛋白质-蛋白质交互界面的表示,然后通过图神经网络使用学习到的表征进行验证。作者的方法在测试集上达到了0.91的准确率,超过了现有的基于GNN的方法。作者在一个基准数据集上展示了方法的有效性,结果显示它是验证蛋白质-蛋白质界面里一个有希望的解决方案。

在计算研究中,蛋白质通常通过其序列和/或结构来表示。三维结构捕捉原子的排列并提供有关功能位点和交互面的详细信息。深度学习方法被用来分析这些3D结构和蛋白质的主序列,并从中提取有意义的特征。卷积神经网络(CNNs)通过对蛋白质结构的局部区域应用滤波器来学习分层表示。然而,CNN并不具有旋转不变性,并且需要数据增强来减轻定向敏感性,这会影响训练期间的计算性能。最近,图神经网络(GNNs)因其将蛋白质-蛋白质界面表示为旋转不变图而变得流行,从而克服了这些限制。GNN基础的研究将蛋白质建模为图,其中节点代表氨基酸或原子,边表示空间、序列和/或功能关系。随着基于图的深度学习方法的进步,基于图的蛋白质表征学习也受到了广泛关注,因为它能够捕捉蛋白质-蛋白质界面上的复杂交互和结构依赖性。通过利用多视图对比学习和自监督任务,有效捕捉了蛋白质重要的几何特征。transformer也被用来研究蛋白质-蛋白质界面,并通过结合Vision Transformer架构与混合和多注意力网络来区分类似天然的蛋白质复合物和错误构象。
本研究提出了一种基于图的方法,称为ProInterVal,用于借助广泛的训练数据集,利用学习到的界面表示来验证蛋白质-蛋白质界面的生物学相关性。作者的方法结合了一种新颖的图对比autoencoder和transformer来学习表示,并随后用GNN模型验证界面。与之前的研究类似,作者的模型包含一个多视图对比层,用于捕捉蛋白质内相关亚结构之间的相似性,以及一个边信息传递层,用于捕捉涉及残基及其邻近残基的不同交互之间的相互依赖性。ProInterVal通过利用学习到的蛋白质-蛋白质界面表示,使其与其他模型区分开来。使用少量手工制作的特征仅限于生成用于表示学习的输入图,因为蛋白质-蛋白质界面的验证完全依赖于学习到的表示。作者的方法在测试集上显示出0.91的准确率,超过了当前基于GNN的方法的表现。
数据部分
本研究使用的蛋白复合物数据来源于PDB,使用的数据集包含534203个样本,来源于三篇论文使用到的数据集:DeepInterface、GNN-DOVE、DeepRank-GNN。表1展示了DeepInterface中各数据来源的分布。本研究中,作者将正样本界面定义为PDB中存在的蛋白复合物的界面。而负样本界面定义为已对接但是CAPRI(Critical Assessment of Predicted Interaction)分数很低的蛋白复合物里的界面,CAPRI的评判标准是Fnat(fraction of native contact)值低于0.1,LRMSD(ligand root-mean-square deviation)大于10埃,iRMSD(interface root-mean-square deviation)大于4埃。
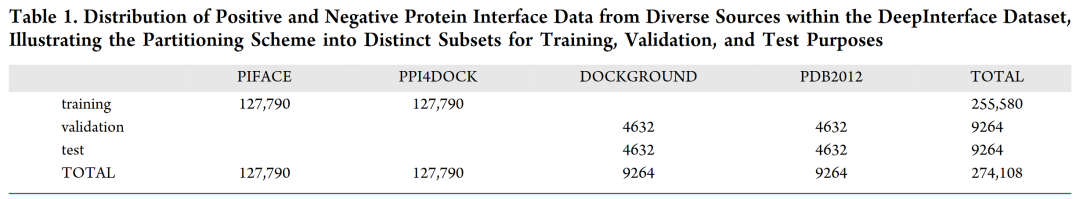
表1
研究中最开始的步骤为抽取出蛋白复合物中的PPI界面。复合物中,每个结合伙伴由一个蛋白反应位点,其中有些残基直接与其他伙伴的反应位点接触,而有些相邻的残基尽管没有直接接触,但也在结合过程中起到了作用。通过比较不同链上残基之间的距离,来评估残基的接近程度。如果两个残基之间的距离满足阈值要求(小于0.5埃),作者就认为它们是接触残基(contact residue),而在接触残基同一链中离接触残基6埃的残基定义为邻居残基(neighboring residue)。接触残基与邻居残基的并集为蛋白复合物的界面区域。
作者将界面区域建模为图,其中残基表示为节点,反应表示为边。每个节点的维度为30,由以下特征组成:残基类型、极性、残基电荷、单体和复合体形态中的相对可及表面积(relASA)、基于知识的配对势(PP)、主链二面角(φ 和 ψ)。
模型部分
模型输入为包含两个链的蛋白复合物。模型的整体流程为:最开始,抽取界面(interface)并将其转换为图表征形式(如“数据部分”所述);随后,利用一个基于图的对比autoencoder与transformer架构来获得图的隐空间表示;最后基于学到的表征,利用GNN模型为两个链生成一个反应分数。
作者这里的图对比学习参考了Protein representation learning by geometric structure pretraining文献中提到的三种边类型:Sequential edges、Radius edges、K-nearest neighbor edges。唐建团队的GearNet工作基于蛋白结构进行预训练也采用了这三种边类型。具体来说,本研究中:如果两个残基在序列中的距离小于3,两个残基间构成一条Sequential edge;如果两个残基之间的距离小于10埃,两个残基间构成一条Radius edge;最后,给每个残基都找出欧几里得距离最近的k(=10)个残基,之间构成一条K-nearest neighbor edge。
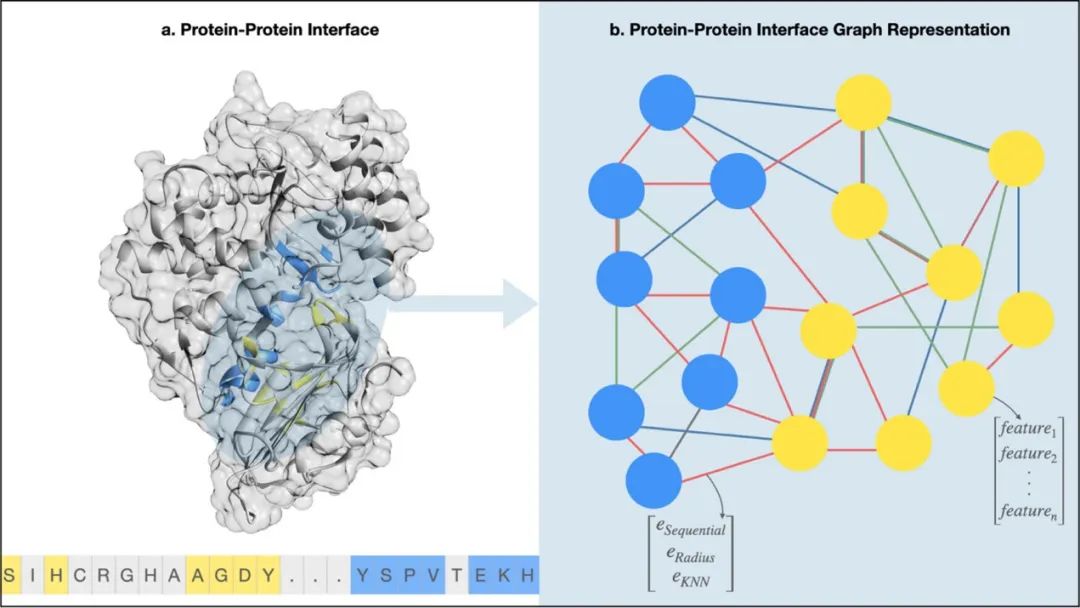
图 1
图1展示了蛋白复合物的3D结构(界面残基高亮表示)与序列,及其图表征形式。其中节点有着节点相关的特征(维度30),而边取值范围为{0, 1},表示某一边类型是否存在。
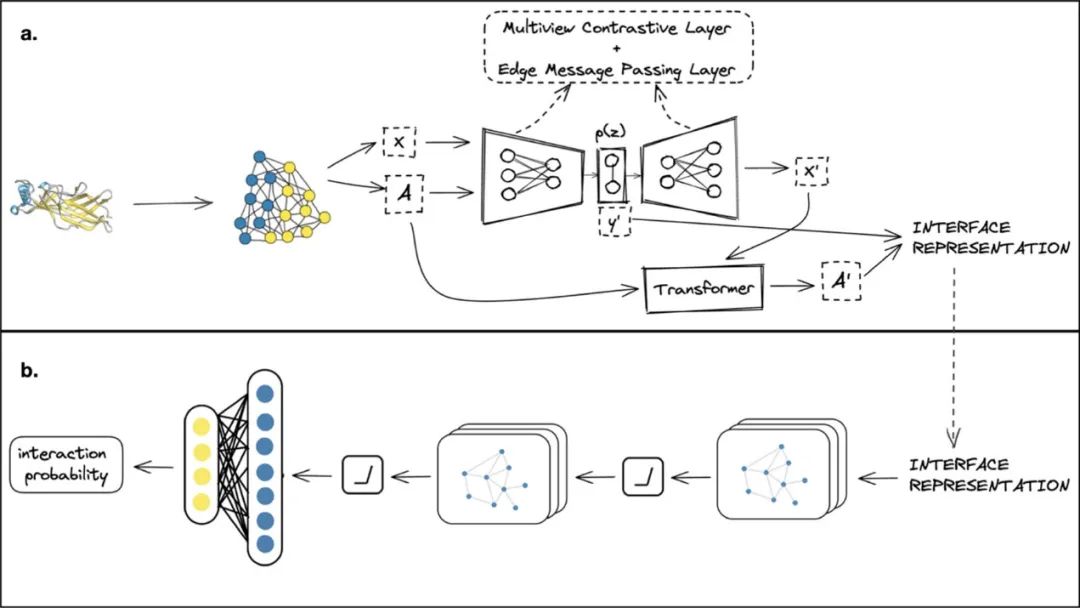
图 2
图2展示了作者本篇提出的模型。encoder输入节点矩阵X和邻接矩阵A,将其编码为连续隐空间表征z。而给定隐空间中的一个点,decoder生成一个输出节点特征矩阵X'。另外,利用解开的隐空间,模型预测出图属性预测y'。
autoencoder中加入了一个多视角对比学习层(Multiview Contrastive Layer)和边信息传递层(Edge Message Passing Layer)来增强模型性能。多视角对比学习包含了两个裁剪操作:子序列裁剪(subsequence cropping),子空间采样(subspace cropping)。其中子序列裁剪为随机采样蛋白质氨基酸序列片段,抽取出对应的子图;子空间采样为随机选定一个中心残基,将特定距离阈值内的残基纳入构成一个子图。随后,对输出的子图不进行变换(identity操作)或随机移除图中一定比例的边(random edge masking操作)。边信息传递中存在一个关系图(也叫线图,line graph),其中图中的每个节点代表原图中的一条边。使用RGCN网络对线图进行学习。
Transformer接受邻接矩阵A和预测的节点特征矩阵X’,生成预测边A‘。生成阶段,transformer顺序地往图里添加边。每一步中,transformer输入包括预测边的邻接节点特征以及先前预测的边。Transformer目标函数包括四个损失函数:L_n代表重建节点特征的损失;L_KL代表输入图与隐空间表征的KL散度损失;L_h代表预测图属性的均方差损失;L_t代表重建边特征的交叉熵损失。各自的权重为0.2,0.3,0.3,0.2。
消融实验

表 2
如表2所示,使用多视角对比学习可以大幅提升模型性能,而在加入边消息传递后模型性能进一步提升。
PPI验证性能评估
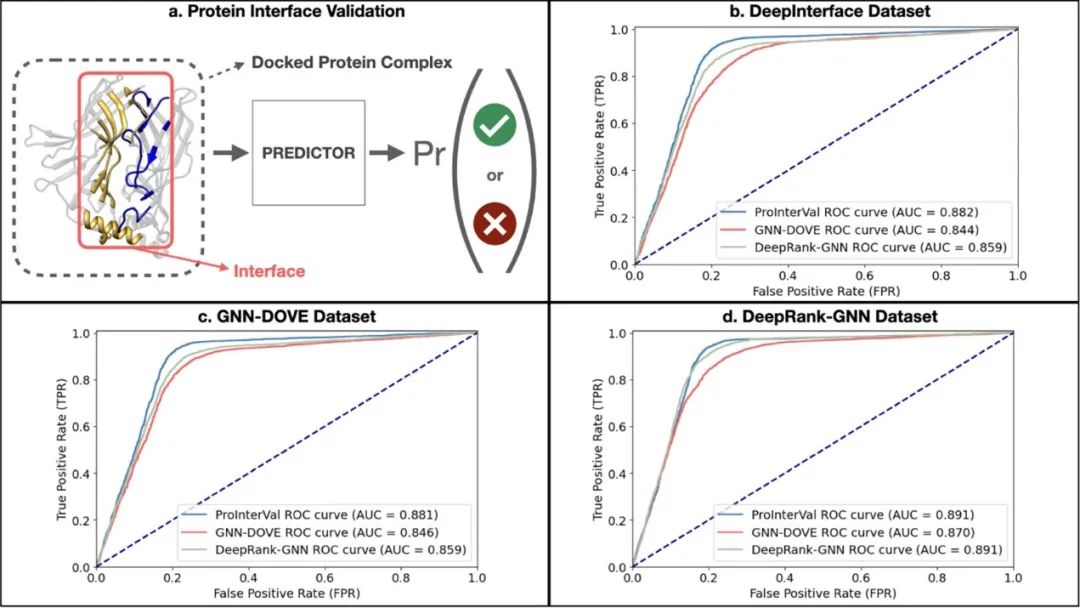
图 3
作者这里将ProInterVal方法与两个sota方法(GNN-DOVE和DeepRank-GNN)进行性能比较。为了公平的对比,三个模型方法都在三种数据集上训练与测试。从图3可以看出,ProInterVal在三个数据集上都超过了其他的模型。
t-SNE分析
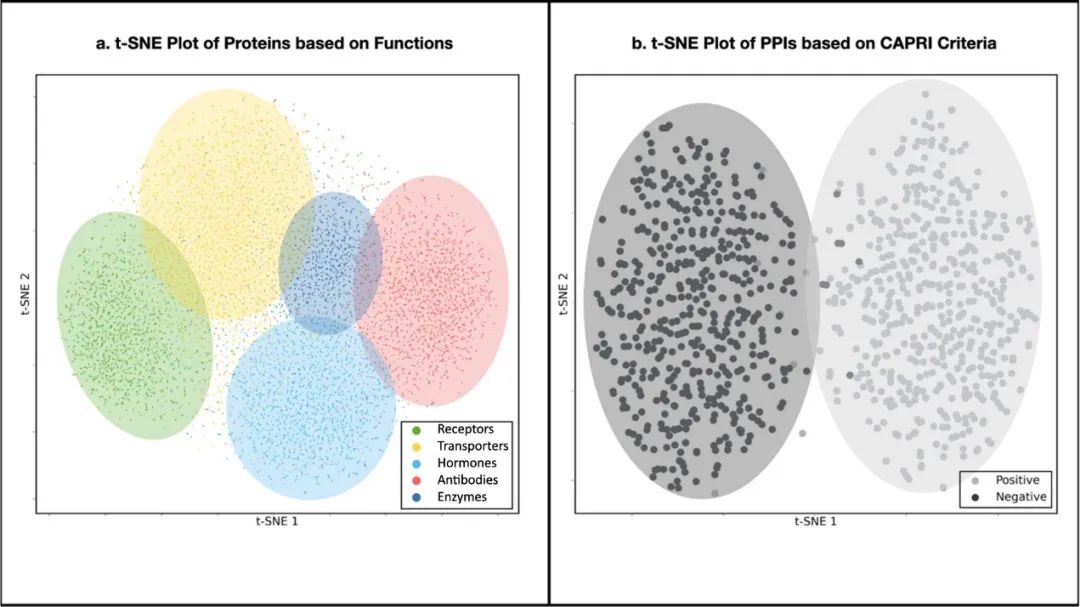
图 4
图4展示了对蛋白功能簇与正负界面点的t-SNE分析。这些可视化结果展示了ProInterVal方法在理解蛋白反应与生物学意义上的潜力。如图4a所示,蛋白质根据嵌入空间中的功能类别进行聚类,表明ProInterVal在某种程度上捕获了蛋白质的功能指纹。为了更深入分析模型学习到的界面特征,基于CAPRI准则随机选取了500个正样本界面和500个负样本界面。从图4b中可看出,正负样本界面在特征空间中完全地分隔开了,证明ProInterVal模型有效地学习到了PPI验证的鲁棒特征。
正样本界面排序
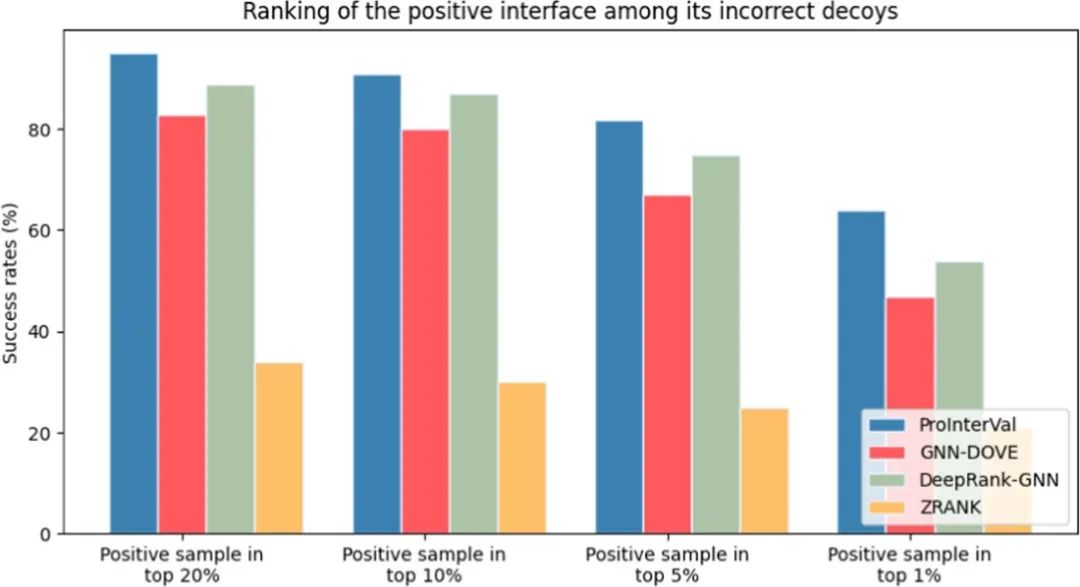
图 5
为了全面评估模型,作者进行了一个排序分析:让模型从100个不正确的对接构象(负样本)中辨别一个天然的界面(正样本)。具体来说,作者从DeepInterface测试集中随机选取了100个蛋白复合物正样本,对于这100个复合物,利用GRAMM-X蛋白对接工具生成300个decoy,然后根据CAPRI准则选取出其中的100个decoy。这样就组成了100组(1个正样本+100个负样本)。
随后,作者使用ProInterVal,GNN-DOVE,DeepRank-GNN,ZRANK模型对这些结构进行评分与排序。top 10%指的是在对应排行列表中接近正样本的几面放在了前10%。图5展示了排行结果。模型在top1%中成功率为63%,top 90%中成功率为90%,超越了其他的模型。
生物与晶体界面分类
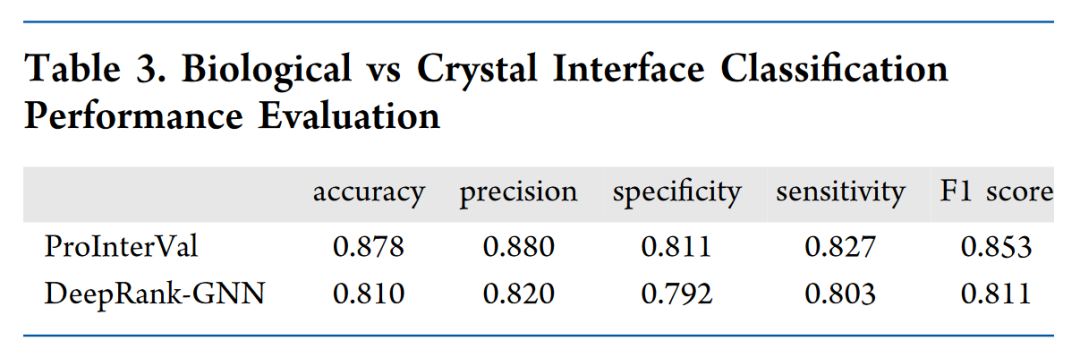
表 3
X射线晶体学长期以来一直是确定蛋白质三维结构的主要实验方法,并且自2022年以来仍然是最广泛使用的技术。虽然X射线结构通常是可靠且高质量的,但错误的结构并不少见。常见的错误包括残基错误配位到电子密度图中以及由于晶体堆积而形成的合成寡聚体的存在。后者可能导致误导性的结论,并可能阻碍准确的研究发现。因此,开发工具来标注晶体学二聚体作为可靠的生物或非生物(晶体)界面是至关重要的。
本章节中,作者利用DC数据集来评估ProInterVal辨别生物与晶体结构的性能。具体来说,为了训练和评估 ProInterVal,作者使用了包含来自 MANY 数据集的 5739 个二聚体的数据集。训练集包括了 80% 的二聚体,而剩余 20% 的二聚体构成了验证集。网络训练了 50 轮,选取了在验证集中损失最低的模型作为最终模型。随后,这个模型在 DC 数据集上进行了测试,该数据集包含 80 个生物界面和 81 个具有相似界面面积的晶体界面。从表3可看出,ProInterVal在测试集上达到了接近88%的准确率,88%的精度,85%的F1 score,各项指标都超越了DeepRank-GNN。
编译 | 黄海涛
审稿 | 王建民
参考资料
Ovek, Damla, Ozlem Keskin, and Attila Gursoy. "ProInterVal: Validation of Protein–Protein Interfaces through Learned Interface Representations." Journal of Chemical Information and Modeling 64, no. 8 (2024): 2979-2987.
内容中包含的图片若涉及版权问题,请及时与我们联系删除

评论
沙发等你来抢