
DRUGAI
今天为大家介绍的是来自Degui Zhi团队的一篇论文。准确预测蛋白质-配体结合亲和力在药物发现中至关重要。现有方法主要是不涉及对接的,当无法获得结晶的蛋白质-配体结合构象时,这些方法并未明确考虑蛋白质与配体之间在原子级别的相互作用。作者引入了一个框架——折叠-对接-亲和力(FDA)。该框架折叠蛋白质,确定蛋白质-配体的结合构象,并从三维蛋白质-配体结合结构预测结合亲和力。实验表明,FDA在DAVIS数据集中的表现超越了现有的最先进的无对接模型,展示了明确模拟三维结合构象对提高结合亲和力预测准确性的潜力。

大多数现有的结合亲和力预测方法利用机器学习方法,但它们通常不考虑结合姿态,即无对接,因此忽略了配体与蛋白质之间的原子级相互作用,如图1(a)所示。在这些模型中,蛋白质主要表示为氨基酸序列或蛋白质接触图,而配体则用SMILES字符串或分子图表示。这些无对接方法通常作为黑箱模型运行,缺乏结构上下文。虽然它们可以以端到端的方式轻松训练,但这些无对接方法可能缺乏通过对接方法提供的分子相互作用的可解释性和详细洞察力,可能导致不够充分的预测。当共晶的三维结构可用时,在开发基于对接的方法方面取得了显著进展,如图1(b)所示,这些方法明确考虑了原子级相互作用,利用三维卷积神经网络或交互图神经网络来预测结合亲和力。当在收集了高分辨率的晶化三维结合结构PDBBind数据集上测试时,这些基于对接的模型可以胜过它们的无对接对手,这种优越性取决于三维结合结构的可用性。然而,在没有高分辨率结合结构的情况下,基于对接的模型是否仍能保持其优势?本工作旨在回答这一问题。
鉴于AlphaFold和其他方法已经彻底改变了蛋白质折叠领域,获取三维蛋白质结构比以前容易得多。同时,基于深度学习的蛋白质-配体对接模型已被开发出来,当提供特定的蛋白质和配体信息时,可以更容易地生成结合构象,其准确性正在赶上基于计算的分子动力学对接。这些进展为在没有高分辨率晶体三维结合结构的情况下预测结合亲和力开辟了新的方法。
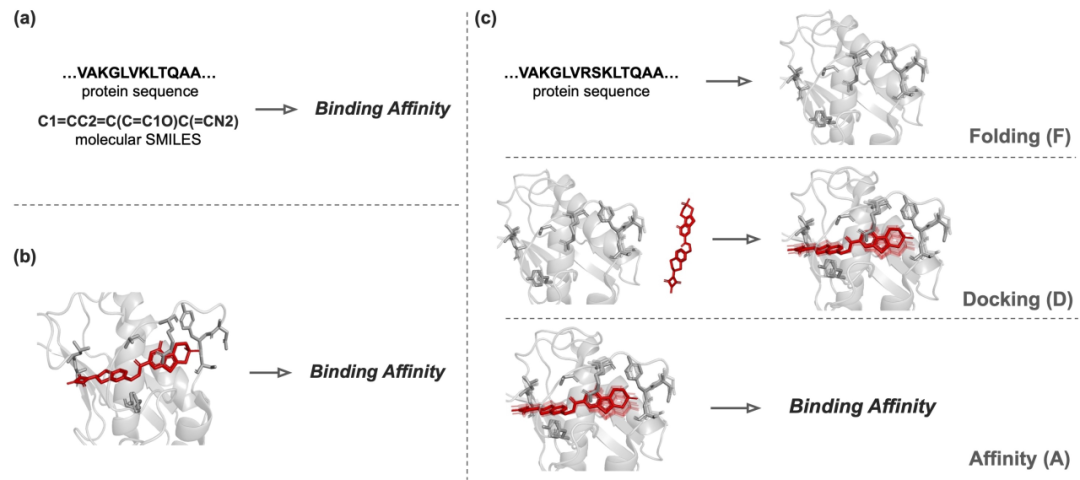
图 1
模型部分
折叠-对接-亲和力(FDA)框架包括三个组成部分,如图1(c)所示:
(i) 折叠:从蛋白质氨基酸序列生成三维蛋白质结构。
(ii) 对接:将配体对接到生成的蛋白质结构上。
(iii) 亲和力:从预测的三维蛋白质-配体结合结构预测结合亲和力。
FDA框架具有个别可替换的组件,使其能够适应各种折叠、对接、亲和力预测方法。作者使用ColabFold(v1.5.5)作为对接组件来生成apo蛋白结构,通过MMseq2生成的MSA特征和模板结构作为输入信息。后端模型架构是Alphafold 2 multimer(v3)。作者对每个结构进行了三次预测循环,并为每个蛋白质生成了五个结构。排名最高的结构随后使用Amber进行了弛豫处理,该过程在OpenMM(7.7.0)中实现。作者使用基于评分的扩散模型DiffDock(v1.0)作为对接组件。对于每个蛋白质-配体对,作者抽样了十个结合姿态。DiffDock包括两部分:一个用于配体结合姿态的扩散生成模型和一个信心评分模型。前者通过生成平移和旋转矩阵以及表示配体扭转角的标量来抽样各种结合姿态。此外,信心评分模型评估生成模型抽样的结合姿态的质量。生成模型和信心模型之间的一个关键区别是生成模型仅考虑蛋白质结构的α-碳,而信心评分模型则考虑蛋白质的所有重原子。作者直接使用预训练的DiffDock模型来生成和随后排名对接姿态。
此外,作者整合了GIGN提出的亲和力预测器,根据排名最高的生成结合姿态预测结合亲和力。GIGN包括三个基本组成部分:结合口袋区域内的蛋白质图、配体图和蛋白质-配体互动图。此外,在图消息传递阶段,模型将共价相互作用和非共价相互作用分开处理,以增强学习节点表示的有效性。
亲和力预测比较
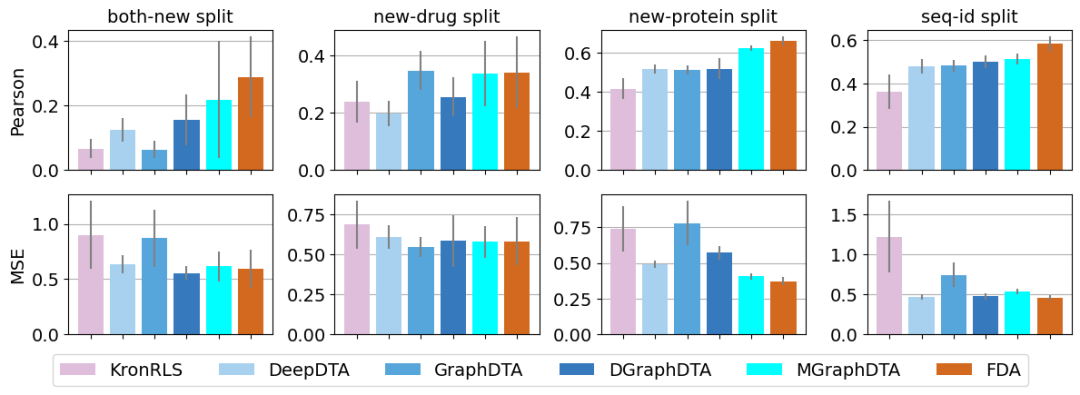
图 2
根据实验结果,FDA模型在图2所示的四个不同的测试集中通常优于其他无对接模型。特别是,在最具挑战性的任务——“全新”分割中,FDA模型展示了其卓越的性能。这种分割涉及测试集中的蛋白质和配体不在训练集中,凸显了该模型的泛化能力。这些发现强调了作者方法的优势,即明确考虑蛋白质-配体结合构象,在提高模型泛化能力方面相比其他无对接方法更为有效。此外,在FDA框架内,与结晶的全体结构相比,使用ColabFold生成的apo结构在亲和力预测性能上带来了意外的提升。
消融实验
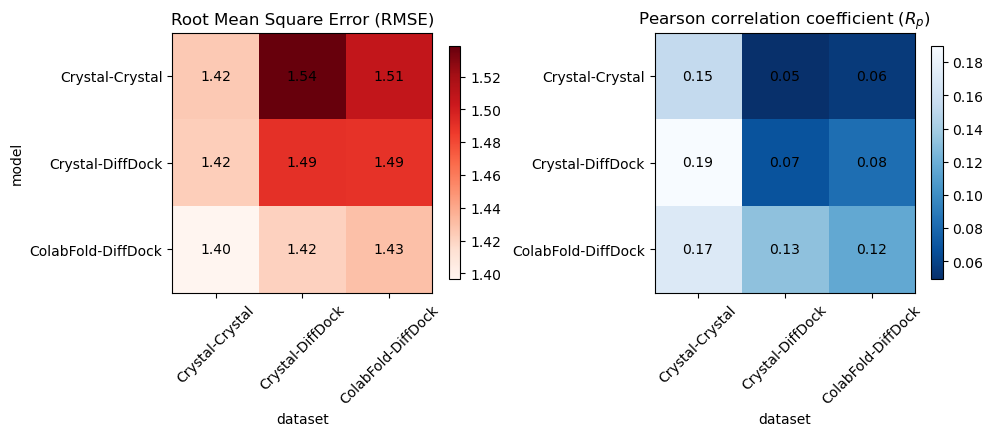
图 3
在使用DAVIS数据集进行的亲和力预测基准测试中,作者测试了FDA方法和其他无对接方法,结果表明FDA方法通常优于这些方法。然而,由ColabFold和DiffDock引入的偏差对最终亲和力预测的影响仍不清楚。为了解决这个问题,作者设计了具有三种不同情景的消融实验:
晶体结合姿态(晶体-晶体):
蛋白质-配体结合结构完全通过实验确定,ColabFold和DiffDock没有引入偏差。
使用DiffDock的全体晶体蛋白(晶体-DiffDock):
在这种情况下使用全体晶体蛋白作为蛋白质结构,并利用DiffDock确定配体的结合姿态。
使用DiffDock的Apo ColabFold(ColabFold-DiffDock):
在这种情况下,作者使用Apo ColabFold蛋白质结构,并再次利用DiffDock寻找配体的结合姿态。
三种不同模型在各种测试集上的性能如图3所示。最初,作者假设使用晶体-晶体数据集训练的模型可能在晶体-晶体测试集上表现出最佳预测性能(由最浅的颜色表示),而预测ColabFold-DiffDock数据集的ColabFold-DiffDock模型可能表现最差(由最深的颜色表示)。这一假设基于由ColabFold和DiffDock引入的潜在错误,该错误可能影响模型的性能。相反,在预测晶体-晶体数据集的ColabFold-DiffDock模型中观察到最低的均方根误差(RMSE),而在预测晶体-晶体数据集的晶体-DiffDock模型中发现了最高的皮尔逊相关系数(Rp)。此外,如图3所示,在三个测试集上,ColabFold-DiffDock模型的表现通常优于其他两种情况。
编译 | 于洲
审稿 | 王建民
参考资料
Wu M H, Xie Z, Zhi D. Protein-ligand binding affinity prediction: Is 3D binding pose needed?[J]. bioRxiv, 2024: 2024.04. 16.589805.
内容中包含的图片若涉及版权问题,请及时与我们联系删除

评论
沙发等你来抢