
今天为大家介绍的是来自北京大学信息工程学院、AI4S平台中心主任陈语谦教授团队发表在人工智能旗舰期刊IEEE Transactions on Pattern Analysis and Machine Intelligence(IF=23.6)的论文,博士生杨梓铎为第一作者。该论文从归纳偏好的角度探讨了深度学习模型在蛋白质-配体亲和力(Protein-Ligand Binding Affinity, PLA)预测任务中的泛化能力和可解释性。归纳偏好是指在深度学习模型中为了更好地进行学习和泛化而引入的假设或偏好。归纳偏好通过限制模型的假设空间,使其在有限的数据上更容易找到合适的模式,从而提高模型的泛化性能。模型的泛化能力及可解释性,往往取决于所使用的归纳偏好在多大程度上能够准确描述待解决的任务。因此,在PLA预测任务中,所采用的归纳偏好应符合物理化学规则,以更好地描述蛋白质-配体间的相互作用,从而提高模型的泛化能力和可解释性。

背景介绍
近年来,深度学习技术已在辅助药物设计中发挥关键作用,尤其是在预测蛋白质-配体结合亲和力方面的应用,加快了从庞大的化学空间中筛选出潜在药物的过程。根据是否考虑复合物的3D结构信息,深度学习方法可分为两大类:不依赖于3D结构的方法以及基于3D结构的方法,如图1所示。不依赖于3D结构的方法通常采用字符串形式的输入,例如使用SMILES来表征配体,使用1D序列(如氨基酸序列)来表征蛋白质。还可以使用2D图来表示配体,但这些表征方式均不涉及3D结构信息。不依赖于3D结构的方法忽略了蛋白质和配体之间的三维相互作用,直接从1D和2D表征中挖掘与PLA预测相关的模式,这些方法在某些情况下可能达到较好的预测结果,但无法保证所挖掘的模式与物理化学规则相符。
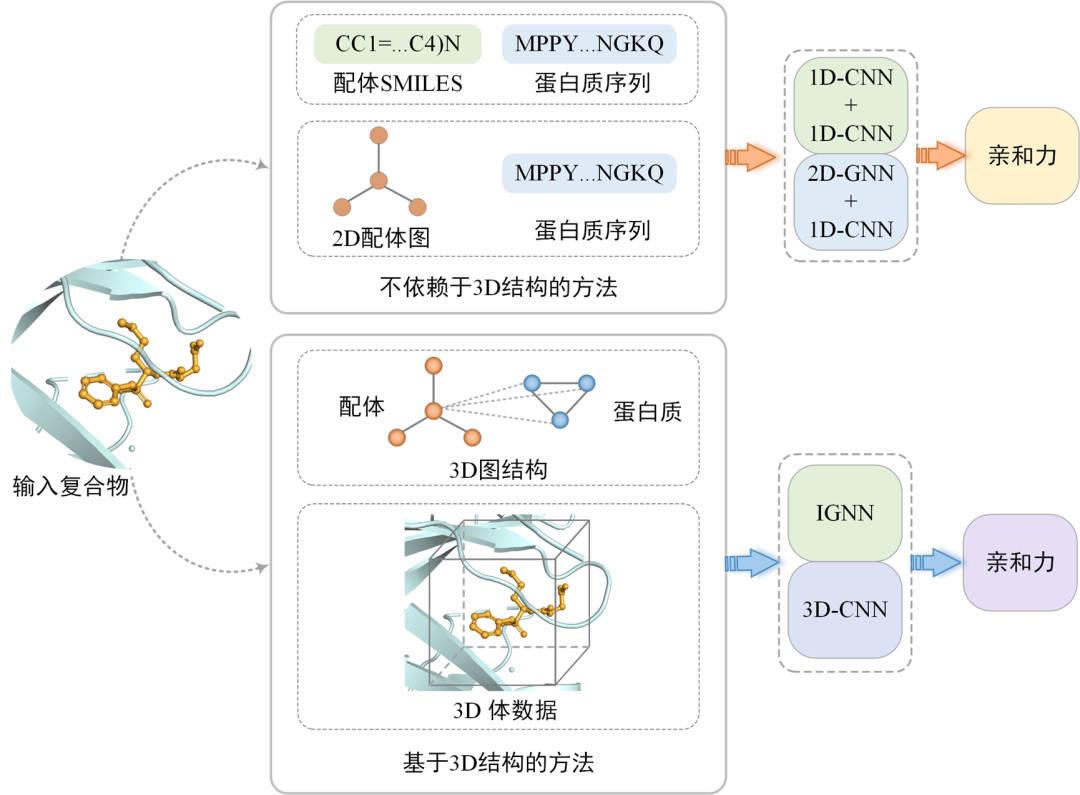
图1. 基于深度学习的PLA预测方法
基于3D结构的PLA预测方法直接利用蛋白质-配体复合物的空间结构和相互作用信息作为输入,能够提供更全面的先验知识。这个特点避免了模型从一维输入中推导三维特征的步骤,使得模型可以更直观地学习分子间的相互作用。这些方法通常将复合物表示为3D网格或3D图结构,然后采用3D-CNN或3D-GNN来提取与结构和相互作用相关的特征。近期的研究已表明3D图结构是一种更高效的表征方式。3D图结构将原子表示为图结构的节点,将化学键表示为图结构的边,而复合物的结构信息则通常由原子对之间的距离来表征。
3D图神经网络(3D-GNN)是一种适合处理3D图结构的深度学习模型。尽管3D-GNN在提高PLA预测的准确性上取得了显著进步,该领域仍面临以下三个挑战:
大多数3D-GNN方法将复合物建模为同质图,即不区分配体和蛋白质原子,也不区分它们之间的相互作用。然而,这种假设忽略了一个重要的事实,即非共价相互作用和共价相互作用在数量上并不匹配。这导致在消息传递过程中,数量较少一方的信息容易被数量较多一方的信息淹没,如图2(a)所示。
大多数3D-GNN模型采用瓶颈架构,即输入的图结构在经过多个图卷积层后,模型使用全局池化将图表示压缩为单一向量,如图2(b)所示。然而,这个过程可能会导致3D结构信息的丢失。
一些3D-GNN模型,如PIGNet(DOI: 10.1039/D1SC06946B),将PLA视为蛋白质-配体原子间相互作用力的总和。虽然这在一定程度上缓解了瓶颈架构导致的结构信息丢失问题,但同时又引入了隐藏偏差。这种偏差主要来源于求和过程中的累积误差,由于每一对蛋白质-配体原子间相互作用力的预测本身可能存在误差,这些误差在计算总相互作用力时会累积,并遵循特定的分布。例如,对于一个复合物,如果非共价相互作用数量较多,那么预测得到的总相互作用力倾向于偏大,但这种趋势与实际情况并不相符。
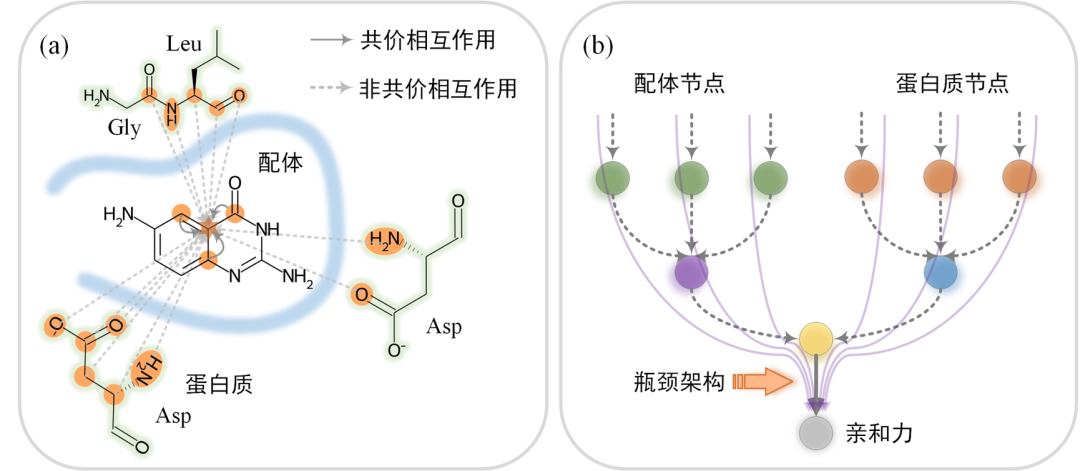
图2. 现有3D-GNN模型的设计思路
方法
为了解决以上问题,论文引入了一种基于相互作用的归纳偏好,包括两个核心假设:第一,蛋白质-配体复合物的结构可以通过异质图来更合理地表示,其中明确区分蛋白质与配体的节点及其相应的边;第二,亲和力的预测值可以被分解为蛋白质与配体之间非共价相互作用力的预测值的累积。这种基于相互作用的归纳偏好具有两个优势:一方面,它通过引入预定的物理规则来缩小假设空间,即减少学习算法必须搜索的假设空间的大小,有助于模型在有限的训练数据上学习到能够更好地泛化到新数据的规律;另一方面,确保了模型对遵循相同物理原则的新数据具有良好的泛化能力。
为了实现该归纳偏好,文章提出了一个可解释的异质相互作用图神经网络(EHIGN)用于PLA预测。EHIGN的核心设计包括如下四个方面:
将复合物建模为异质图,包括配体内部、蛋白质内部、配体与蛋白质间及蛋白质与配体间四种类型的原子间相互作用,如图3(a)所示。
EHIGN使用异质相互作用图神经网络(HIGN)从四种类型的原子间相互作用学习节点表示。HIGN使用四个独立的GNN,包括两个共价相互作用图神经网络和两个非共价相互作用图神经网络,独立地对四种类型的相互作用进行处理,从而避免非共价相互作用信息在消息传递的过程中被淹没。
EHIGN将亲和力的预测值分解为蛋白质-配体原子间非共价相互作用力的预测值之和,如图3(b)所示。这一策略不仅缓解了由于瓶颈架构而导致的结构信息丢失问题,而且提高了模型的可解释性。
EHIGN通过一个可学习的偏差纠正项抵消了隐藏偏差(如图3(b)所示),从而提高了模型的泛化能力。
EHIGN的设计理念与自解释神经网络相似,它不直接预测最终结果,而是预测一系列有意义且易于理解的概念。这些可解释的概念是输入与输出之间的中间变量,模型可以基于这些概念输出最终的预测结果。其关键之处在于如何证明这些概念是可解释的。例如,EHIGN将亲和力的预测值分解为非共价相互作用力的预测值之和,需要证明这些非共价相互作用力的预测值具有物理意义,这是验证模型自解释能力的关键。尽管先前的研究已经尝试将亲和力的预测值分解为非共价相互作用力的预测值之和(DOI:10.1039/d1sc06946b,DOI: 10.1021/acs.jcim.0c00026),但这些研究并未全面探讨这种策略的可解释性,也未深入探讨由该策略所引起的隐藏偏差问题。
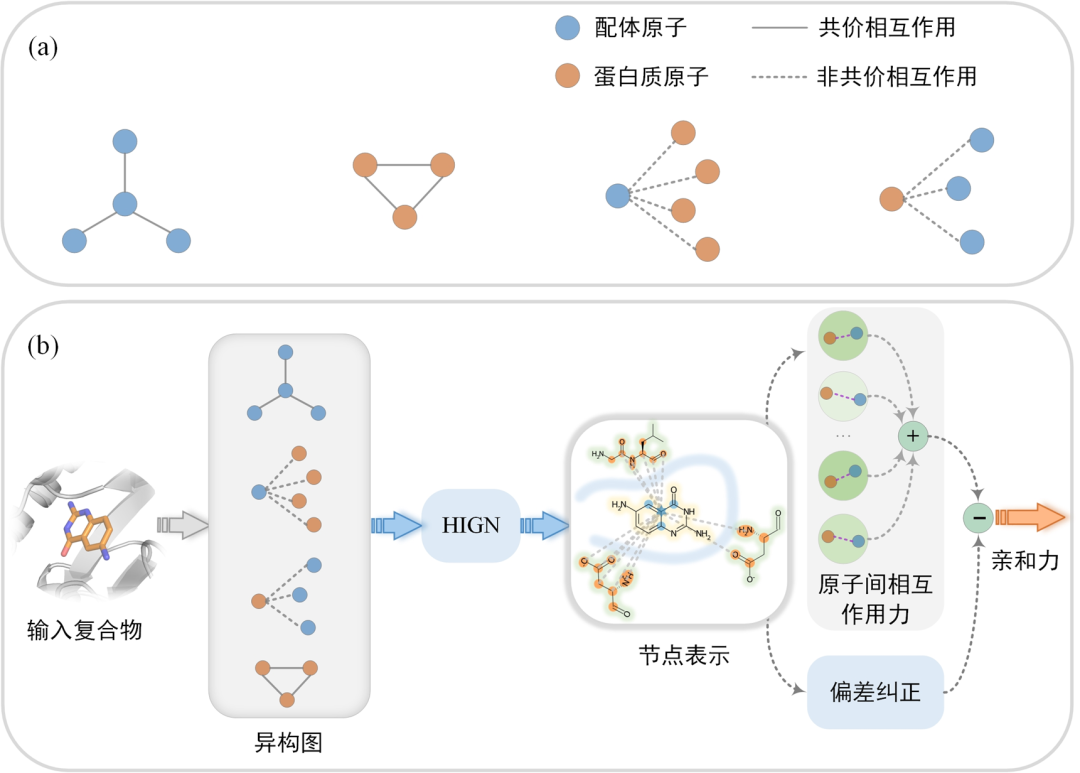
图3. EHIGN的设计理念
模型性能评估
文章首先在PDBbind 2013核心集(N=107)、2016核心集(N=285)、2019保留集(N=4366)以及CSAR-HiQ数据集(N=47)上评估了EHIGN的性能。对比的基线模型包括5个不依赖于3D结构的模型和11个基于3D结构的模型。所有模型均使用相同的训练集(N=11904)、验证集(N=1000)和测试集。实验结果表明两点:第一,基于3D结构的方法性能显著优于不依赖于3D结构的方法。第二,EHIGN在所有测试集中均取得了最佳性能。当基于配体骨架或基于蛋白质序列相似性进行数据集进行划分时,EHIGN仍然取得了最优结果。此外,文章还对EHIGN的虚拟筛选能力进行测试,EHIGN的表现明显优于现有的代表性方法。
模型归纳偏好对模型性能的影响
EHIGN的良好性能主要源自于更合理的模型假设。为了探究基于相互作用的归纳偏好及偏差纠正项对模型性能的影响,文章设计了一系列的消融实验,涉及EHIGN的几种变体,具体如下:
变体1:使用同质图作为输入,并采用图1(b) 所示的瓶颈架构来预测PLA。变体1未考虑基于相互作用的归纳偏好中所做的两个假设。
变体2:仅考虑第一个假设,即异质图假设,但模型仍然遵循瓶颈架构。
变体3:仅考虑第二个假设(亲和力分解假设)和偏差纠正,这意味着输入为同质图。
变体4:同时考虑了两个假设,但未引入偏差纠正项。
消融实验的结果如表1所示。对比变体1和变体2的结果可证明异质图假设的有效性。对比变体4与变体2的实验结果可以证明亲和力分解假设的有效性,其中变体4在PDBbind 2013和2016核心集上的性能显著优于变体2。然而,在包含更多样本的PDBbind 2019保留集上,变体4的性能略低于变体2,这一性能上的降低可归因于隐藏偏差的存在,而此偏差可通过偏差纠正项进行有效缓解。
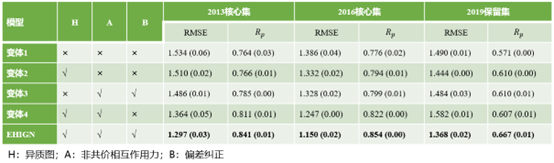
表1 EHIGN及其4种变体在2013核心集、2016核心集和2019保留集上的性能对比
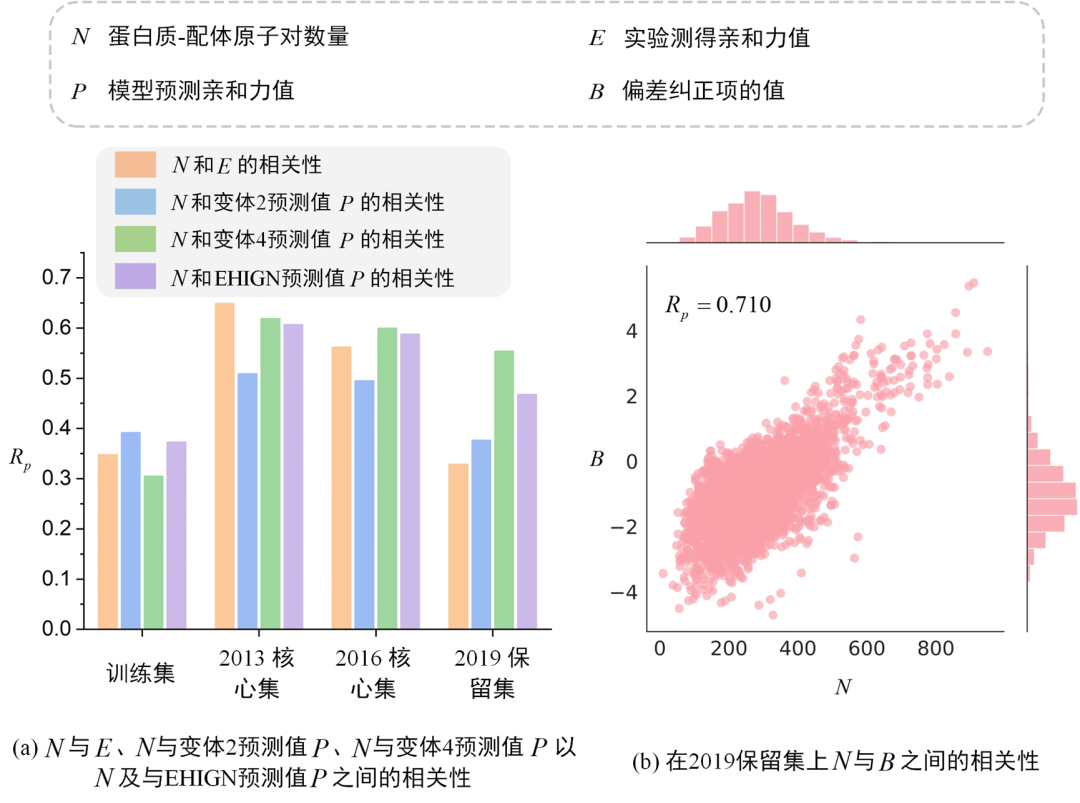
图4. 偏差纠正项的有效性验证
隐藏偏差的来源及偏差纠正项的有效性验证
将PLA的预测值视为蛋白质-配体原子间非共价相互作用力的预测值的累积会引入累积误差,导致模型预测值与蛋白质-配体原子对数量呈正相关性。为了解释这一问题,我们设蛋白质-配体原子对数量为N,实验测量得到的亲和力值为E,模型预测的亲和力值为P,而偏差纠正项的值为B。图4(a)展示了N、E与P之间的关系。在训练集与2019保留集上,N与E的相关性分别为0.348和0.329,表明它们之间存在弱相关性。然而,在2019保留集上,N与变体4的预测值P的相关性为0.554,这暗示变体4可能过度依赖于N与E之间的相关性进行预测,从而导致其在2019保留集上的性能下降。尽管N与E之间存在弱相关性,但过分依赖这一信息可能会导致模型泛化能力的降低。
另一方面,在2019保留集上,N与变体2的预测值P之间的相关性为0.377,这表明变体2未过度依赖N与E之间的弱相关性。由于变体2采用的是瓶颈架构,受此启发,可学习偏差纠正模块也采用了瓶颈架构,通过对蛋白质-配体成对节点表示进行求和,可以得到一个与非共价相互作用数量相关的表示,这有助于捕获隐藏偏差。与变体4相比,在2019保留集上,N与EHIGN的预测值P的相关性降至0.468,表明偏差纠正模块在一定程度上能够缓解隐藏偏差,从而提升模型的泛化性能。
为了进一步验证偏差纠正模块设计的合理性,我们分析了2019保留集上蛋白质-配体原子对数量N与偏差纠正项的值B之间的相关性,结果如图4(b)所示。N与B之间的相关性达到了0.710,这一相关性证明了偏差纠正模块能够有效地识别隐藏偏差。
模型可解释性
为了验证EHIGN的推断过程与生物化学机理的相关性,我们基于PDBbind v2016数据集探究了模型预测的蛋白质-配体原子间相互作用力与原子间距离的关系。结果表明,配体原子与其周围所有蛋白质原子的总非共价相互作用力与最近蛋白质原子之间的距离存在较强的负相关关系,相关系数为-0.620。类似地,蛋白质原子与其周围所有配体原子的总非共价相互作用力与最近配体原子之间的距离也存在中等强度的负相关关系,相关系数为-0.506。这些结果表明存在一种趋势:当配体原子与其周围蛋白质原子之间的最短距离减小时,施加在该配体原子上的非共价相互作用力的预测值有增大的趋势,这与物理现象相符。这一结果证明了EHIGN具备一定程度的自解释能力,同时也表明了其可以保留空间结构信息。
此外,针对模型预测的蛋白质-配体非共价相互作用力,我们进行了可视化分析,以探究距离-亲和力、构象-亲和力和亚结构-亲和力之间的关系,结果分别如图5、图6和图7所示。其中表示施加在配体原子上的非共价相互作用力。通过可视化结果,我们得出以下三个结论:第一,预测的相互作用力的强度与氢键的存在密切相关(见图5);第二,相互作用力的模式随靶标的不同而变化(见图6);第三,亲和力预测值的差异主要由配体中不同亚结构的贡献度差异引起(见图7),这表明通过EHIGN预测得到的每个亚结构的亲和力贡献度可以为配体优化提供物理意义上的洞见,展示了EHIGN在配体设计优化中的潜力。
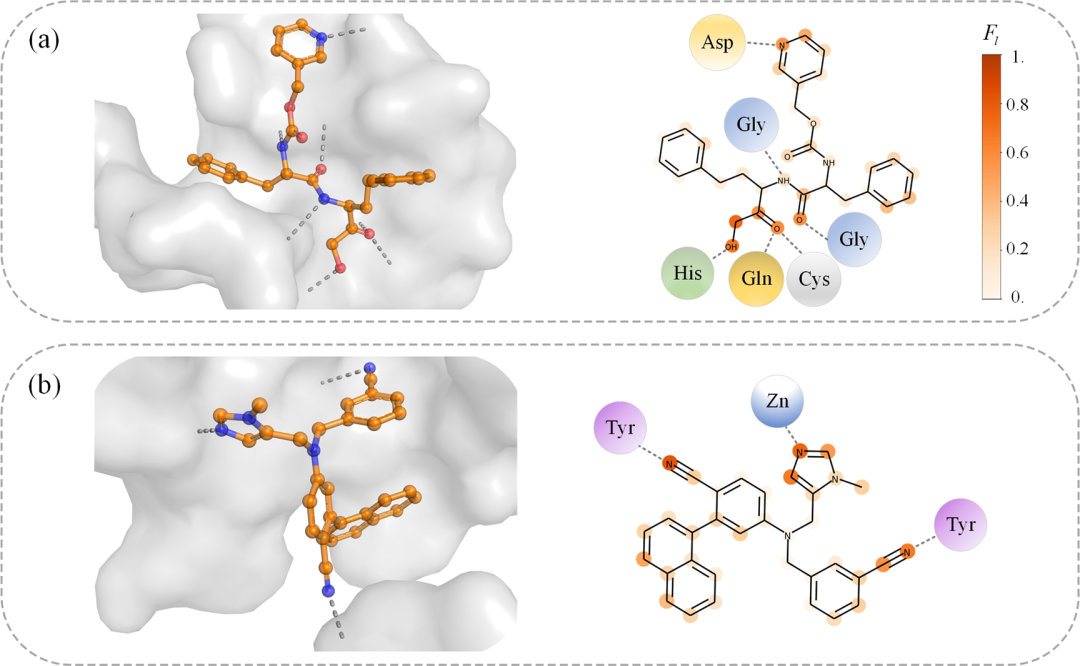
图5 蛋白质-配体结合模式的3D和2D可视化,复合物的PDB ID分别为:(a)1me3(b)1nl4
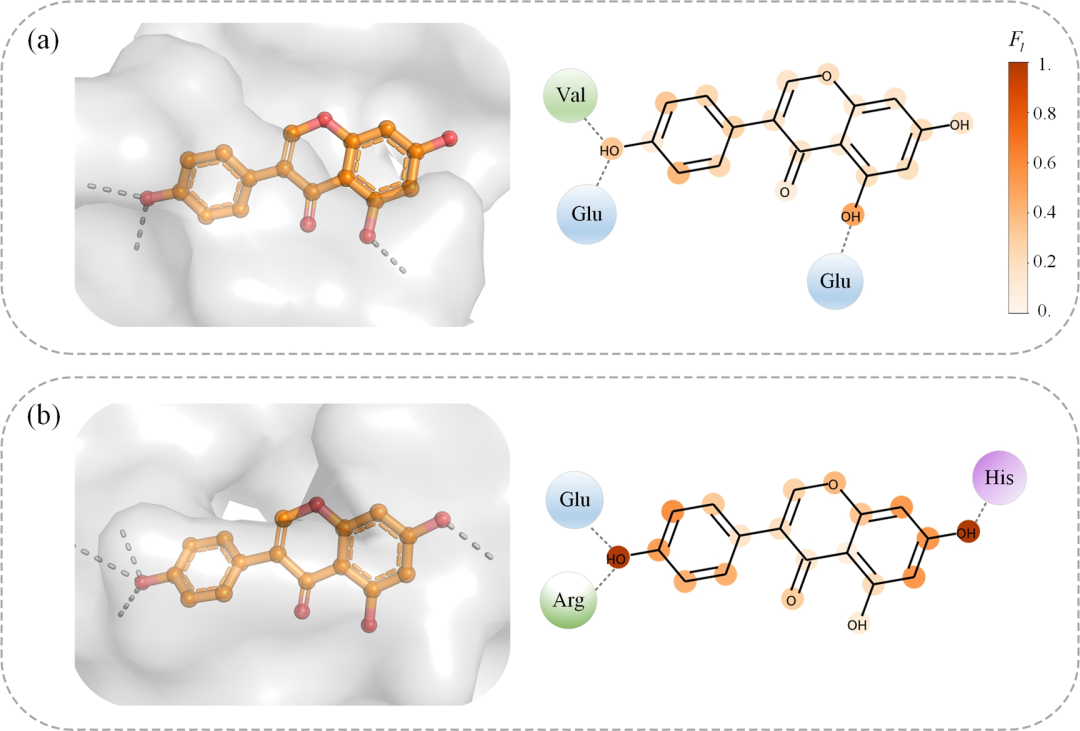
图6 蛋白质-配体结合模式的3D和2D可视化,展示了芹菜素与两个不同蛋白质的结合模式:(a)死亡相关蛋白激酶1(PDB ID: 5auz)(b)雌激素受体1(PDB ID: 1x7r)
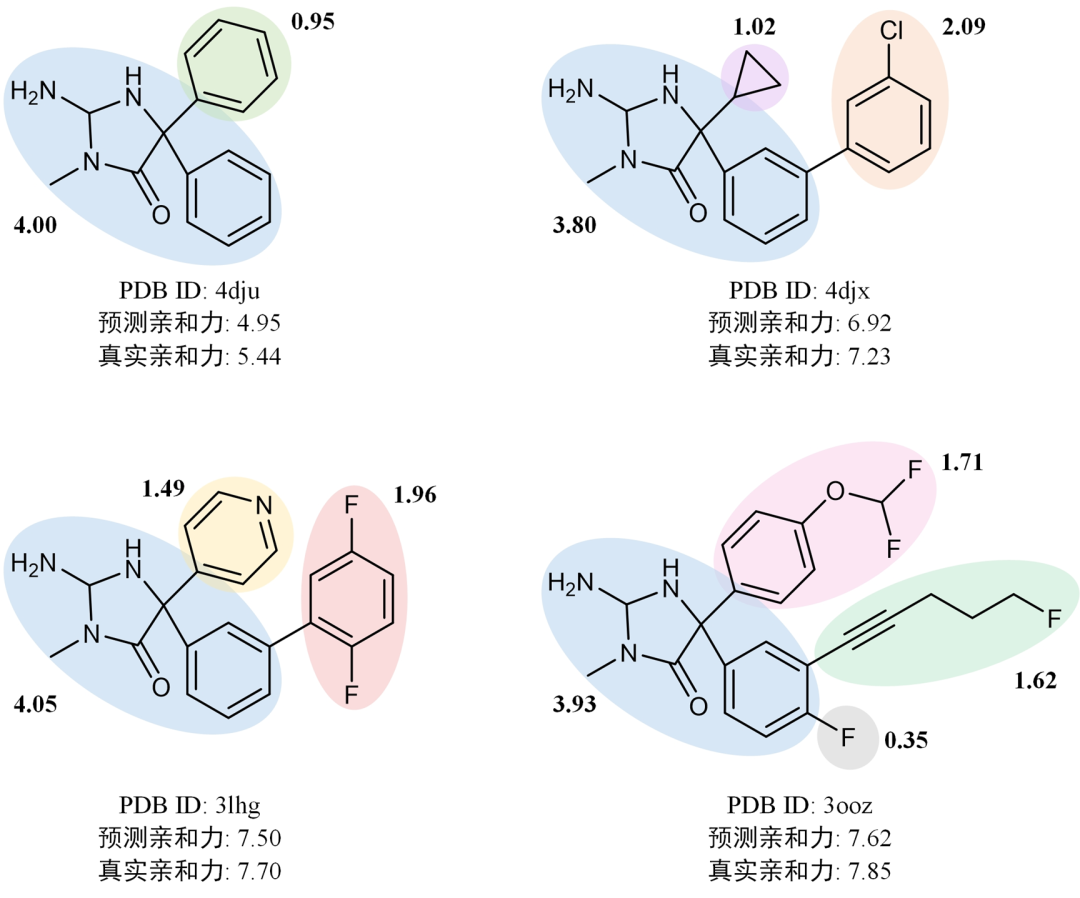
图7 配体中不同亚结构对亲和力预测值的贡献度可视化,配体中共有的最大公共亚结构以蓝色区域显示
结论
文章从归纳偏好的角度探讨了深度学习模型在PLA预测任务中的泛化能力和可解释性,指出在PLA预测任务中,所采用的归纳偏好应符合物理化学规则,以更好地描述蛋白质-配体间的相互作用,从而提高模型的泛化能力和可解释性。为了实现这一目标,文章提出了一种基于相互作用的归纳偏好,通过引入两个核心假设来约束模型的假设空间,确保神经网络能够学习到与蛋白质-配体结合模式相关的特征。EHIGN在PLA及虚拟筛选场景中均表现出色。进一步地,通过对距离-亲和力、构象-亲和力和亚结构-亲和力之间关系的定量和定性分析,证明了基于相互作用的归纳偏好可以指导模型学习与物理事实相符的知识,从而赋予EHIGN一定程度的自解释能力。
参考资料
Yang Z, Zhong W, Lv Q, et al. Interaction-Based Inductive Bias in Graph Neural Networks: Enhancing Protein-Ligand Binding Affinity Predictions From 3D Structures[J]. IEEE Transactions on Pattern Analysis and Machine Intelligence, 2024.
文章链接:https://ieeexplore.ieee.org/abstract/document/10530021
代码:
PLA预测:https://github.com/guaguabujianle/EHIGN_PLA
虚拟筛选:https://github.com/guaguabujianle/EHIGN_SBVS
其它基线模型:https://github.com/guaguabujianle/GIGN
内容中包含的图片若涉及版权问题,请及时与我们联系删除

评论
沙发等你来抢