
DRUGAI
今天为大家介绍的是来自Wojciech Matusik团队的一篇论文。近年来,分子发现的研究主要集中在小型药物分子上,导致许多同样重要的材料设计应用缺乏足够的技术支持。这些应用通常依赖于更复杂的分子结构,并且这些结构往往是通过已知的子结构精心设计出来的。作者提出了一种数据高效且可解释的模型,用于表示和推理此类分子,该模型使用图语法明确描述了以基序为设计基础的层次化设计空间。作者提出了一种新颖的表示形式,即在设计空间上的随机游走,这有助于分子的生成和性质预测。作者证明了在性能、效率和预测分子的可合成性方面,该方法相较现有方法具有明显优势,并且提供了关于该方法化学可解释性的详细见解。

以性质为驱动的分子发现是一项具有巨大社会潜力的应用,在近年来机器学习社区的大量研究中得到了反映。然而,大多数研究都集中在小型药物分子上,而许多更复杂的分子类别却被大大忽视了。用于气体分离膜或光伏等应用的材料,这些对可持续未来至关重要的材料,通常具有与典型药物分子显著不同的分子结构分布。此外,这些设计和使用案例的特定性以及实际实验的高昂成本,常常导致数据和标签的缺乏;例如,包含约300个分子或更少的数据库并不罕见。因此,材料科学尚未完全利用机器学习方法的潜力。作者专注于这些具有挑战性的数据集,这些数据集包含在多个不同的实际应用场景中应用的复杂分子,这些分子包含官能团和结构基序。
作者的目标是以数据高效且可解释的方式表示和推理分子。特定领域的数据集通常表现出不同的基序和官能团,这些作为结构先验信息用于作者的分子表示。先前的研究表明,结构先验对于需要数据效率的应用非常有利。作者提出了一种新颖的方法,用于分子发现,该方法适用于更复杂的分子和低数据场景,并基于上述见解。作者的想法是从一组专家定义的基序开始,并在基序空间上学习一个上下文敏感的语法。本文的创新之处在于对这种语法的表示和学习。
一种可解释的、基于语法的分子表示和高效的学习
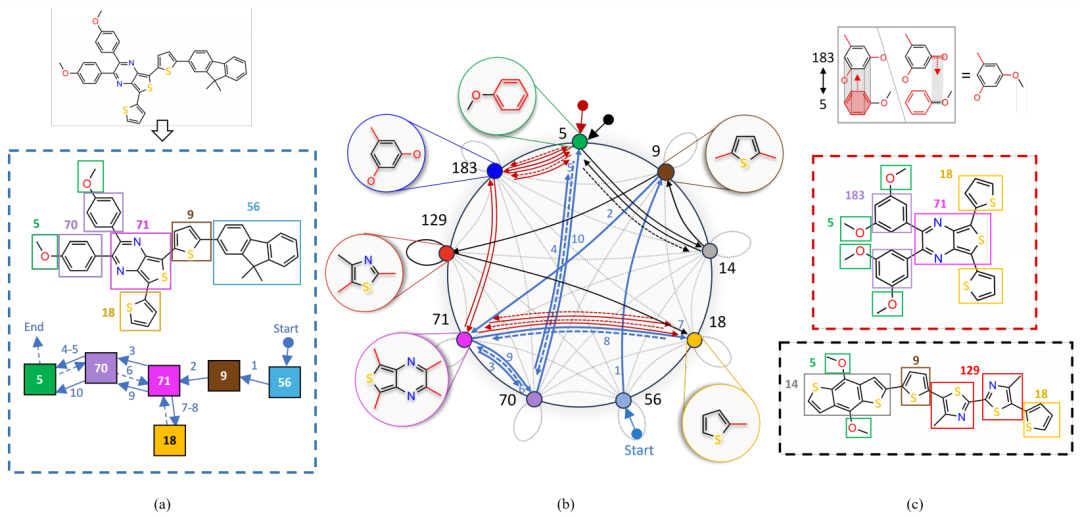
图1:随机游走表示法的说明
作者介绍了一个基于语法的分子表示和高效学习方法。该方法采用图语法,包含一组预定义的分子基序和一组转换规则,基序可以通过自动生成或手动策划获得,并通过转换规则相互连接形成完整的分子结构。
具体而言,作者定义了一种随机游走语法,确保生成分子的过程是渐进的,每一步都会将新的子图附加到现有图上。该语法使用一个紧凑的基序图来实现(见图1b),其中节点代表基序,边表示转换规则的应用。
该方法的两个主要创新点为:
分子被表示为在连接子图上的随机游走(见图1a),这种表示明确、紧凑且具有可解释性。
基于给定训练数据集学习上下文敏感的语法,通过优化边权重来参数化转移概率,从而影响分子表示并促进规则学习。
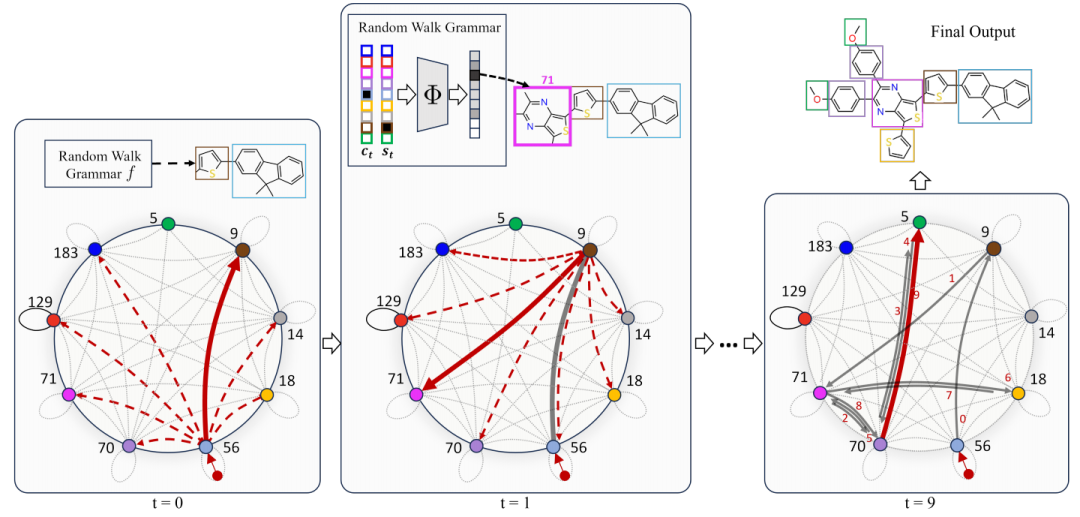
图2:生成过程的说明
如图2所示,为了生成一个分子M,作者将学习到的语法向前应用到随机游走过程中的样本边进行遍历。
实验结果与分析
表1:属性预测结果
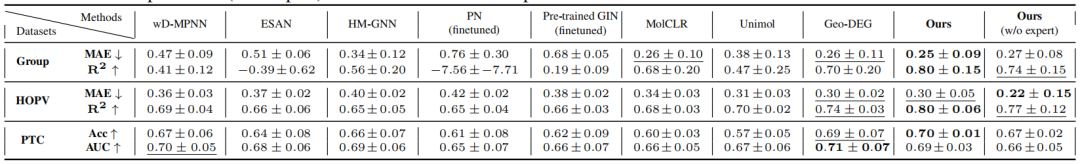
作者的方法在使用专家基序时,在回归数据集Group Contribution和HOPV上分别以0.10和0.06的R²优势超过第二好的方法,并且在PTC数据集上达到了最高的准确率。使用启发式基序时,作者的方法在两个回归数据集上的R²仍然高于Geo-DEG,在PTC数据集上的准确率也在标准差范围内。值得注意的是,在HOPV数据集中,使用启发式基序比使用专家基序和Geo-DEG的平均绝对误差(MAE)显著降低了27%。作者观察到,专家式方法通常能更好地识别启发式方法无法察觉的特殊情况,而启发式方法则更一致。这反映了R²通常比MAE对异常值更敏感。
表2:HOPV(上)和PTC(下)的分子生成结果
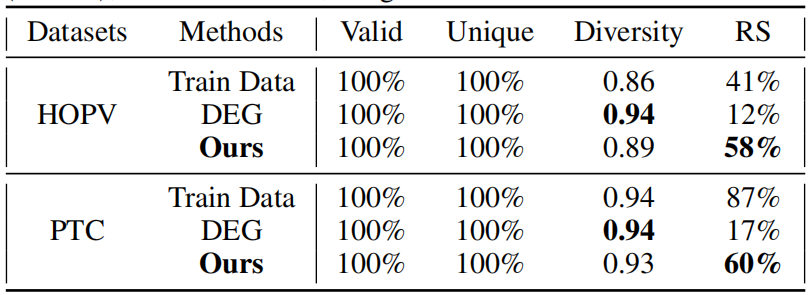
如表2所示,作者的方法比训练数据集生成了更多样化的分子(在HOPV上增加了0.03,在PTC上减少了0.01),并且生成的可合成分子显著多于之前的最先进方法DEG(在两个数据集上都增加了43%)。在HOPV数据集中,作者的逆合成规划器在生成的新分子上找到合成路径的成功率比原始数据集高14%,这对于依赖设计可合成性的实验科学家来说是一个鼓舞人心的消息。
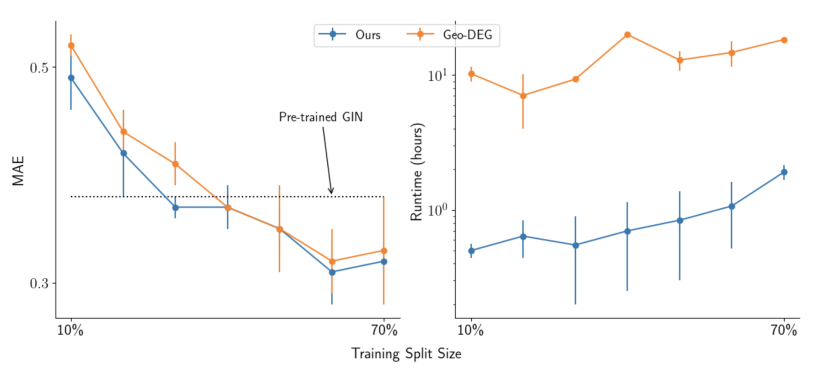
图3:使训练数据集的大小在10%-70%之间发生变化
作者在图3中进行了一个关于训练集划分大小的消融研究,以研究该方法在数据和运行效率方面与Geo-DEG的对比。当训练集从70%减少到10%时,作者的方法在MAE上的表现明显更好。此外,该方法的运行速度提高了一个数量级,突显了在数据效率和运行效率上的提升。
表3:在每个数据集上修复了相同的测试数据
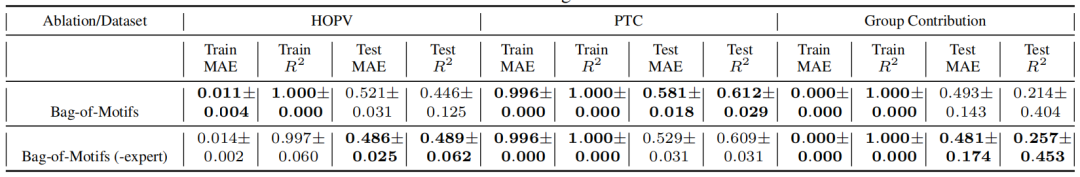
作者去除层次化信息,仅保留基序共现信息。对于每个分子获得一个特征向量,包含a)所有基序的出现次数和b)分子的Morgan指纹。作者在这些特征的基础上训练了一个XGBoost回归器/分类器。如表3所示,该基线模型有足够的能力过拟合训练数据,但无法推广。这表明在缺乏适当表示的情况下,基序出现信息不足以实现泛化。有趣的是,在这种特征化中,专家级基序并不优于启发式基序。这表明在没有包含每个基序细粒度特征的层次化表示的情况下,基序的质量并不重要。
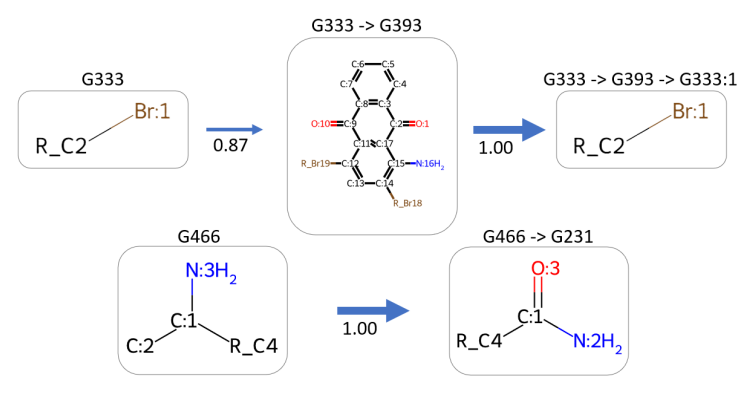
图4:可视化PTC上的两个硬上下文敏感的规则,它们对应于添加卤素基团的设计原则,以进一步提高分子毒性
图4显示了作者的模型如何找到一套设计原则,基于PTC的结构-性质关系用于促进功能分子的合成。考虑到一个三联苯衍生物分子(标记为[‘G333’, ‘G393’]),在加入溴基后(标记为[‘G333’, ‘G393’, ‘G333:1’]),其中间部分G393具有两个对称的酮基和两个连接在芳香环上的溴原子。这种配置显著增强了分子的毒性。此外,通过在芳香环上定位额外的结合位点,模型将两个额外的溴基G333加入到分子中,从而加剧了其肝毒性。在另一个例子中,带有氨基的分子(标记为[‘G466’])在加入一个酮基后(标记为[‘G466’, ‘G231’])发生了转变。在这里,乙酰胺基团中的C=O双键是肝毒性的关键贡献者。
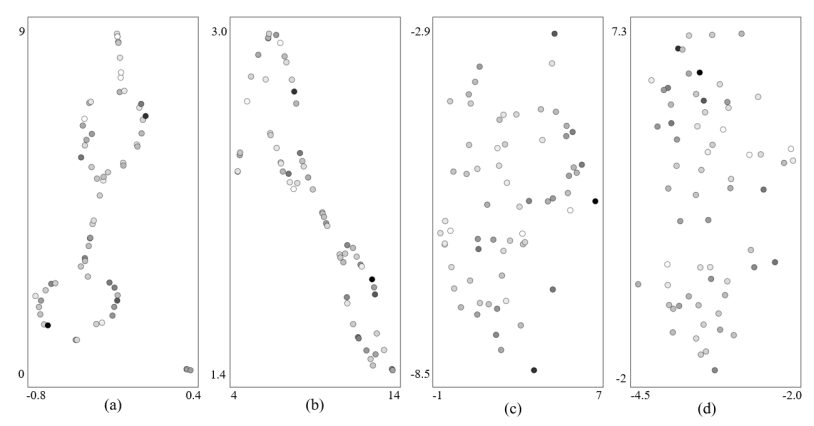
图5:来自最终层的表示
作者的方法在提取视觉上有意义的表示方面独具一格,从视觉聚类中识别出了高HOMO分子进行结构分析(见图5a, b)。
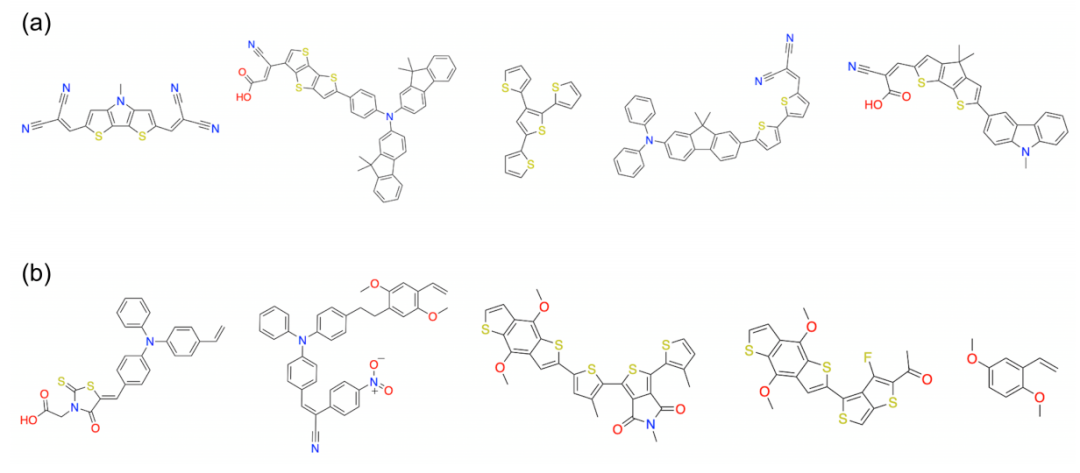
图6:顶部HOMO值化合物的例子,(a)来自上部聚类,(b)来自下部聚类
如图6所示,上部聚类中的分子通常具有促进电子离域的结构,如碳肼腈二腈;而下部聚类中的分子则具有电子供体基团或增加空间位阻的结构,以提高HOMO值。这两个结构特征对应了设计高HOMO值分子的两种主要方法。这些发现有助于寻找具有理想光伏性能的新型分子。
结论
作者将分子表示为在基序图上的可解释的上下文敏感语法上的随机游走,这是一种设计空间的层次抽象。作者在下游性质预测和分子生成任务上的评估表明,这种表示在性能和效率方面结合了定量优势,同时在简单性和可解释性方面具备定性优势。作者设计并执行了一个实际工作流程,通过将分子分解为成熟的功能基团,邀请专家参与,以增强该设计基础和表示,创造专家反馈与表示质量之间的协同效应。未来研究的一个有前景的方向是通过可学习的和/或人类引导的方法与大型语言模型相结合,改进基序的自动提取。
编译 | 于洲
审稿 | 曾全晨
参考资料
Sun M, Guo M, Yuan W, et al. Representing Molecules as Random Walks Over Interpretable Grammars[J]. arXiv preprint arXiv:2403.08147, 2024.
内容中包含的图片若涉及版权问题,请及时与我们联系删除

评论
沙发等你来抢