

DRUGAI
今天为大家介绍的是来自Daniel Jesus Diaz团队的一篇论文。基于AI的蛋白质工程框架使用自监督学习(SSL)来获得用于下游突变效果预测的表示。最常见的训练目标是野生型准确性:在一个序列或结构中屏蔽一个野生型残基,然后预测缺失的氨基酸。然而,野生型准确性与蛋白质工程的主要目标不符,后者是建议突变而不是识别自然界中已存在的氨基酸。作者在此提出进化排名(EvoRank),这是一种结合从多序列比对(MSAs)中提取的进化信息的训练目标,用于学习更多样化的蛋白质表示。EvoRank对应于在MSA引导的概率分布中对氨基酸可能性进行排名。这个目标迫使模型学习蛋白质的潜在进化动态。在各种表型和数据集上,作者证明了EvoRank在零样本性能方面有显著提升,并且可以与在实验数据上进行微调的模型竞争。

AlphaFold的成功开启了蛋白质设计和工程领域深度学习框架的新纪元。大型蛋白质语言模型(如ESM)、结构生成模型(如RFDiffusion、NeuralPLexer)以及基于结构的自监督模型能够加速生物技术的发展,在识别致病变异和用于生物制造的酶工程等应用中具有重要作用。由于生成实验数据的成本高昂,自监督学习(SSL)已成为该领域用于生成蛋白质表示的主要技术。这些方法依赖于屏蔽蛋白质中的残基,然后预测野生型(WT)氨基酸作为SSL训练目标。
对于机器学习指导的蛋白质工程(MLPE),研究人员希望模型能建议蛋白质的突变,而不是仅仅预测野生型。为了解决这一差异,已经提出了几种方法。基于结构的方法通常会调整对数值的温度以偏离野生型。基于序列的方法需要大型蛋白质数据库,并将多序列比对(MSAs)作为额外输入。然而,一个更严重且常被忽视的问题是,野生型准确性的提高可能与下游突变效果的性能不相关。作者在表1中明确展示了这一现象,通过训练一个基于结构的模型以提高野生型准确性,并显示其在热力学稳定性预测方面的下游性能在超过某个野生型准确性阈值后开始下降。

表 1
开发一种自监督学习目标,作为突变效果预测的有效代理任务,仍然是一个关键的未解决问题。因此,作者提出了一种新的自监督训练目标EvoRank,该方法结合了多序列比对(MSAs)中的进化信息,以解决WT-mask SSL的局限性。
MSA软标签损失函数
传统的WT-mask自监督任务中,一个氨基酸会被遮盖掉,然后训练一个网络f使其基于该遮盖掉的氨基酸附近的微环境从而预测回原本的氨基酸,这样一来网络就可以给下游任务提供有效的蛋白质表征。
而作者希望通过自监督学习过程实现以下目标:(1) 避免分布偏向野生型的低熵分布,(2) 从输入的蛋白质结构中结合有意义的进化和生化信息。由于多序列比对 (MSA) 提供了一个强有力的工具来捕捉序列之间的进化关系,作者提出通过MSA软标签损失(公式2)将MSA信息引入自监督学习,其中将野生型的独热编码标签替换为来自蛋白质MSA的分布。
形式上来说,作者并不是训练网络f去预测野生型氨基酸的独热编码标签,而是基于从蛋白质的MSA中得到的概率密度函数,预测以下软标签(式1)。其中,l是20种氨基酸之一,δ是delta函数,MSA(P)表示在UniRef50上通过多序列比对与P最佳对齐的序列集合,Amino(P’, j)表示蛋白质P’在位置j的氨基酸类型。作者将这种分布成为经验氨基酸分布。
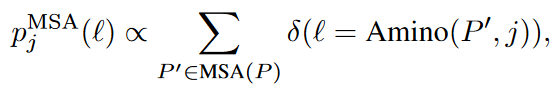
式 1
于是,作者将MSA软标签损失函数定义如式2所示。

式 2
尽管KL散度是经典选择,但它存在模式坍塌的问题。作者尝试使用更丰富的α散度家族中的D(·; ·)。通过应用不同的α值,作者可以调整对MSA中存在的多峰分布的敏感性,并找到对排名最高的氨基酸(通常是野生型)进行过高或过低估计之间的更好平衡。当作者应用反向KL散度或α = 0.5的散度(表2)时,观察到排名顺序略有改善,但总体上前五种氨基酸的系数较低。这表明需要设计更好的损失函数。

表 2
EvoRank:一种新的基于排名的目标函数
为了进一步提升自监督模型的性能,作者重新构建了训练任务,使其更直接地对应于突变预测,并使用排序损失进行训练。与其预测野生型氨基酸类型或软标签,作者设置了一个模型,输入一对“正”与“负”的氨基酸类型a+和a−,并输出它们在经验氨基酸分布中的相对可能性。更具体地,作者定义了相对于(a+, a−)的排序标签如式3。其中代表根据对氨基酸a分配的概率,确保当= 时做出中性预测。
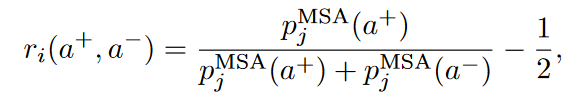
式 3
排名标签(rank label)表示在特定微环境下两种氨基酸在进化上被观察到的相对可能性,如图1所示。
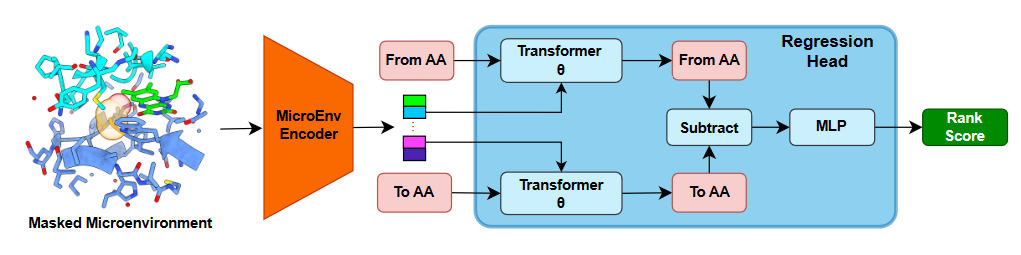
图1
然后作者通过以下损失函数(式4)去训练一个模型f去预测排名标签。其中,a+和a−在所有氨基酸类型上求和,并且D(x, y) = ||x − y||。作者将公式4中的损失称为EvoRank损失或EvoRank训练目标。
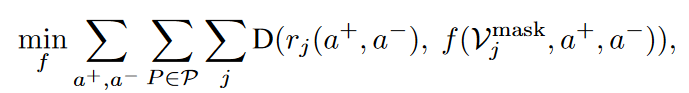
式 4
实际上,作者首先使用MSA软标签损失(公式2)进行训练以初始化参数,然后应用EvoRank损失以进一步提升性能。类似的思想在推荐系统文献中也有应用,这些研究先使用标准预测损失训练模型以初始化参数,然后再使用排名损失进行进一步训练。
零样本热力学稳定性评估
作者使用MSA软标签损失和EvoRank损失重新训练了一个SOTA结构模型。用EvoRank损失训练的MutComputXGT结构模型被命名为MutRank,作者将这个模型称为MutRank。
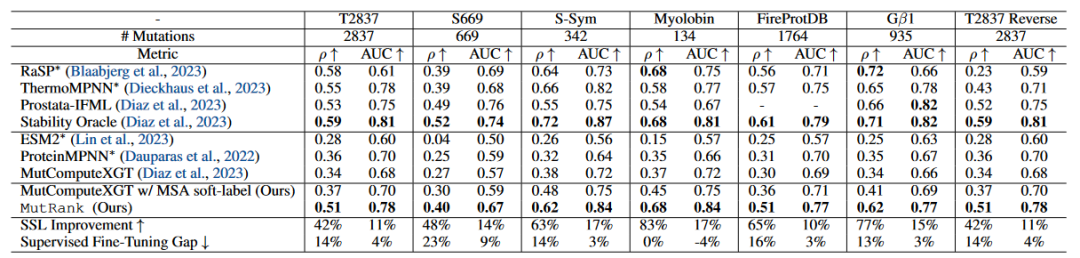
表 3
表3报告了在多个ΔΔG数据集上的不同机器学习框架的零样本皮尔逊相关系数(ρ)和ROC曲线下面积(AUC)的表现:T2837,S-Sym,S669,FireProtDB,Gβ1和肌红蛋白。作者的结果验证了在自监督训练中优先考虑排名顺序对零样本ΔΔG预测的影响。首先,基于MSA的软标签与α散度的结果在皮尔逊相关系数和AUC上均已超过文献中的自监督基线。然后,通过重新构建EvoRank训练目标,作者显著提升了之前文献中最好的零样本模型的表现——在六个数据集上的皮尔逊相关系数和AUC分别平均提升了约64%和约14%。与其WT mask前身MutComputeXGT直接比较,MutRank在皮尔逊相关系数和AUC上分别提升了66%和16%。值得注意的是,与著名的自监督方法ESM2和ProteinMPNN相比,MutRank在六个ΔΔG数据集上的皮尔逊相关系数平均分别提高了约288%和约72%。这些结果展示了MutRank表示在ΔΔG预测中的有效性。
接下来,作者将MutRank与基于结构的框架RaSP和ThermoMPNN以及基于序列的框架Prostata-IFML进行了比较。尽管这些框架在大规模cDNA ΔΔG数据集上进行了明确的微调,但作者的零样本结果也具有竞争力。与SOTA监督框架Stability Oracle相比,作者的零样本皮尔逊相关系数和AUC在六个数据集上的平均值仅低约13%和约3%。总体而言,作者的结果表明EvoRank损失显著缩小了监督微调框架与零样本表示在ΔΔG预测上的差距。
多种表型的零样本评估
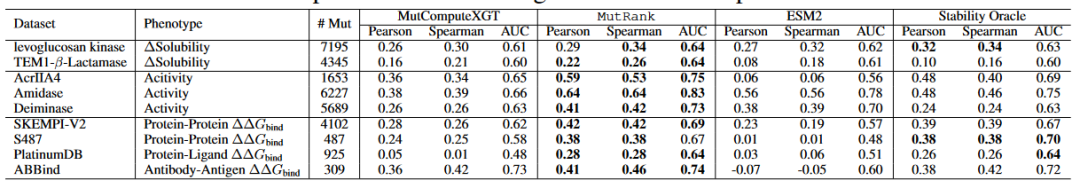
表 4
为了进一步表征MutRank表示的泛化能力,作者评估了其在结合自由能变化数据集和四个DMS数据集上的表现:两个用于溶解度,两个用于活性(表4)。与折叠稳定性不同,后者在可用的公共数据上已有显著增加(Tsuboyama等,2023),而结合自由能变化数据集稀缺,充满了突变类型和标签偏差,并且受到标签噪声的影响。这些挑战使得为这些表型开发监督框架变得困难,并强调了零样本自监督模型的重要性。
对于结合自由能数据集,作者使用了蛋白质-蛋白质界面结合ΔΔG数据集SKEMPIv2,AB-Bind,S487和蛋白质-配体界面结合ΔΔG数据集PlatinumDB。对于溶解度和活性数据集,作者使用了深度突变扫描(DMS)数据集,这些数据集利用高通量筛选或下一代测序作为功能的代理。对于溶解度评估,作者使用了来自Klesmith等的levoglucosan kinase(uniprot id
B3VI55)和TEM1-β-内酰胺酶(uniprot id P62593)的DMS数据集。对于活性评估,作者使用了脂肪族水解酶(uniprot id P11436)、抗CRISPR蛋白AcrIIA4(uniprot id A0A247D711)和卟啉原脱氨酶(uniprot id P08397)的DMS数据集。作者将两个WT掩码自监督学习框架MutComputeXGT和ESM2以及一个监督微调框架Stability Oracle进行了比较。文献方法在结合ΔΔG数据集上的比较结果表明,ESM2在所有指标(皮尔逊和斯皮尔曼相关系数以及AUC)中表现最差,而Stability Oracle表现最好。这些结果是预期的,因为结合自由能(蛋白质之间的相互作用)从根本上与折叠自由能(蛋白质内部的相互作用)相关。ESM2无法看到结合伙伴(蛋白质或配体),必须纯粹依赖于单序列表示。
值得注意的是,MutRank在所有数据集和所有指标上都优于MutcomputeXGT。这表明与其WT掩码前身相比,EvoRank损失提升了所有表型的零样本泛化能力。此外,MutRank在所有数据集和所有指标上都优于ESM2,即使MutRank是一个较小的模型,仅在约23K个蛋白质上训练,而ESM2则在UniRef50上训练。令人惊讶的是,MutRank的零样本性能在几乎所有结合ΔΔG数据集的指标上超过或持平于Stability Oracle的表现(除了S487的AUC)。此外,MutRank在TEM1-β-内酰胺酶溶解度数据集和三个活性数据集上显著优于Stability Oracle。Stability Oracle在TEM1-β-内酰胺酶数据集上的表现低于其预训练表示MutComputeXGT。这一发现突显了EvoRank损失的优越表型泛化能力,并展示了监督微调如何以牺牲其他表型为代价来提升一种表型的性能。
最后,作者强调MutRank在蛋白质-配体界面结合ΔΔG数据集PlatinumDB上的显著改进:与MutComputeXGT相比,MutRank将皮尔逊相关系数和AUC从0.05和0.48(表明一个随机分类器)提升到0.28和0.64。作者总结,对于活性、溶解度和结合自由能表型,MutRank表示显著提升了MutComputeXGT的WT掩码表示的零样本泛化能力。然而,仍需要进一步评估以更好地理解其在多样蛋白质表型中的泛化能力。
编译|黄海涛
审稿|曾全晨
参考资料
Gong, C., Klivans, A., Loy, J. M., Chen, T., & Diaz, D. J. (2024, March). Evolution-Inspired Loss Functions for Protein Representation Learning. In ICLR 2024 Workshop on Generative and Experimental Perspectives for Biomolecular Design.
内容中包含的图片若涉及版权问题,请及时与我们联系删除

评论
沙发等你来抢