
DRUGAI
今天为大家介绍的是来自北京航空航天大学的吕卫锋团队的一篇论文。设计新分子对药物发现和材料科学至关重要。最近,旨在模拟分子分布的深度生成模型在缩小化学研究空间和生成高保真分子方面取得了令人鼓舞的进展。然而,当前的生成模型仅专注于建模二维键合图或三维几何图,这两者是分子的互补描述符。无法将它们联合建模的能力限制了生成质量的提高以及后续的应用。在这篇文章中,作者提出了一种联合二维和三维图扩散模型(JODO),它生成表示完整分子的几何图,包括原子类型、形式电荷、键信息和三维坐标。为了在扩散过程中捕捉二维分子图和三维几何图之间的相关性,作者开发了一个扩散图转换器(DGT)来参数化数据预测模型,该模型从噪声数据中恢复原始数据。DGT使用一种关系注意机制来增强节点和边表示之间的交互。这种机制与标量属性和几何向量的传播和更新同时进行。作者的模型还可以针对单个或多个量子属性用于逆向分子设计。在全面的无条件联合生成评估中,实验结果表明JODO在QM9和GEOM-Drugs数据集上的表现显著优于基线模型。此外,作者的模型在少步快速采样、逆向分子设计和分子图生成方面也表现出色。作者的代码可以在https://github.com/GRAPH-0/JODO获取。

机器学习在分子科学中的应用日益增加,并且已经产生了显著的影响,例如AlphaFold和新药设计。在分子的分析、设计和模拟的各种应用中,发现具有期望特性的新的分子一直是一个长期的挑战,这对药物和材料设计具有促进作用。为了避免在数量庞大的药理学合理的分子中进行暴力搜索,深度生成模型提供了一种强有力的方法来生成新分子并缩小化学搜索空间。
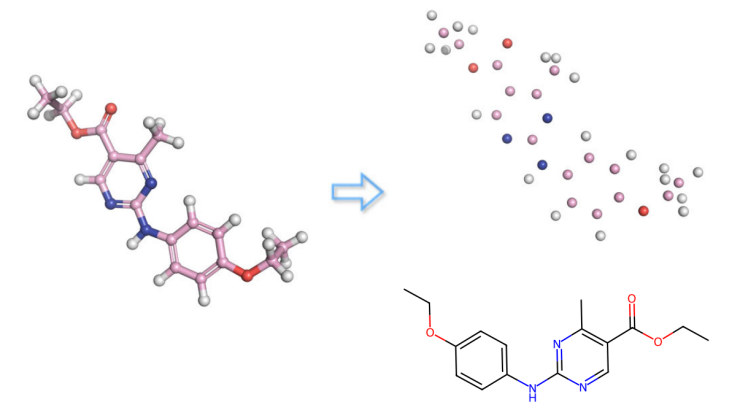
图 1
分子可以用不同的描述符表示,如图1所示,每种表示方式对应不同类型的生成模型。一种常见的表示方式是分子图,它使用节点表示原子,边表示共价键来描述分子的二维键合拓扑结构。这种表示方式对于化学合成、分子动力学模拟等非常方便。因此,许多图生成模型旨在生成真实且有效的分子图。然而,自然分子存在于三维物理空间中,它们的三维结构决定了它们的生物、化学和物理特性。这些特性包括势能面、形状相似性、三维药效团相似性等。二维分子图描述符是一种简化的分子描述符,可能对应多种具有不同特性的空间构象,限制了药物发现中的化学空间探索。分子图生成模型可能会产生不稳定的分子,或者需要额外的模拟来找到低能构象,这妨碍了逆向分子设计和量子特性的优化。此外,结构基础的药物设计等有价值的应用场景也依赖于分子的三维几何结构。
生成从零开始生成原子类型和三维坐标的生成模型,除了二维图生成外,越来越受到关注。通过利用欧几里得空间中的几何对称性(如平移和旋转),这些模型生成现实且稳定的小分子的三维几何结构。然而,仅生成原子的三维位置并不包含分子的键合信息,这带来了一些局限性。一方面,键合信息对于分子生成的质量评估和许多下游应用至关重要。从生成的三维几何结构中确定键的后处理步骤可能会引入错误,并且效果不佳。另一方面,从零开始生成类似于GEOM-Drugs数据集中那些较大药物样分子的高质量三维结构,对于现有的生成模型来说是一个挑战。直观地说,与二维分子图的联合生成可以为几何生成提供额外的有用指导。基于此,作者提出开发几何图(即三维分子图)的生成模型,共同设计二维图和三维几何。这可以提高样本质量,避免键分配的后处理步骤,并促进进一步的应用。
作者的主要贡献总结如下:
1. 作者引入了一种基于联合二维和三维扩散的端到端几何图生成模型,用于完整的分子生成,利用了分子的两种互补描述符,并促进了进一步的应用。
2. 作者开发了一种有效的DGT。它使用一种关系注意机制来在节点和边特征之间交互,捕捉键合图和几何之间的相关性。
3. 作者在QM9和Geom-Drug数据集上的全面评估管道中展示了作者模型的卓越性能。作者的模型还在少步采样、逆向分子设计和分子图生成方面表现出色。
模型部分
在这一部分中,作者介绍JODO,一种用于分子二维键合图和三维几何图的联合扩散模型。作者首先在几何图上定义扩散过程,并引入了等变DGT,以促进生成扩散过程。
A. 联合二维图和三维几何扩散
将分子表示为G = (A, x, h),作者在乘积空间ℝN(N-1)/2*d2*X*ℝN*d1中构建一个连续时间前向扩散过程,以逐渐扰动分子成分及其相关性的分布。这样的过程可以用一个前向随机微分方程(SDE)来描述,其中 t ∈ [0, T]:
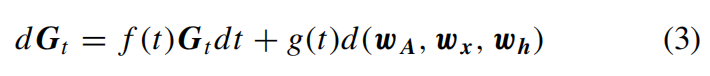
其中(wA, wx, wh)分别是三个空间中的独立标准Wiener过程。作者确保时间和数据空间的连续性,使得可以使用快速采样器等方法。这个前向SDE在(1)中具有线性高斯转移核,可以方便地在任意时间t对Gt =αt G0 +σtεG进行采样,其中εG是乘积空间中的高斯噪声。对应的逆向时间SDE从时间T到0如下所示:
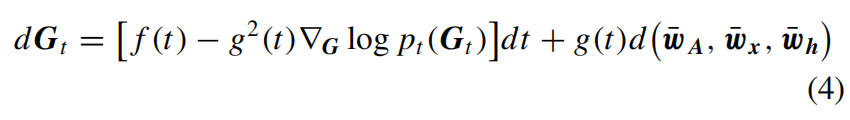
数据预测模型dθ(Gt, G0, log(αt2/σt2))在乘积空间ℝN(N-1)/2*d2*X*ℝN*d1中产生三个输出,分别表示为(dθA, dθx, dθh)。作者通过最小化以下目标函数来优化模型:
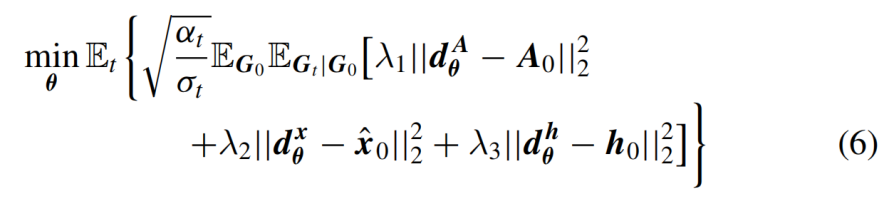
其中λ1, λ2, λ3是三个输出的损失权重。加权项sqrt(αt /σt)使训练目标等效于简单的噪声预测损失。由于dθ应该是SE(3)-等变的,作者遵循使几何监督信号x0与xt等变。作者首先将x0和xt转换到无质心系统,应用Kabsch对齐算法找到最佳旋转矩阵,最终在对齐后获得x0。

作者在算法1中展示了更详细的训练过程。
B. 扩散图Transformer
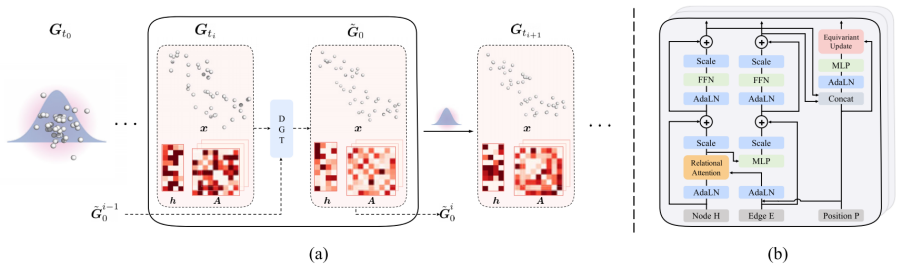
图 2
整体架构如图2(b)所示。扩散模型的生成质量在很大程度上取决于数据预测模型的设计。在几何图生成的背景下,这些模型需要处理三个不同的组件:节点标量特征H、边缘标量特征E和节点的三维坐标P。在扩散过程中,这三个组件会独立地被噪声注入并逐渐失去关联性,这使得数据预测模型难以恢复原始的分子。
为了应对这一挑战,作者提出了DGT,它参数化了dθ(Gt, 0, log(αt2/σt2))。DGT采用了典型的Transformer架构,由多头注意力机制(MHA)、层归一化(LN)和前馈网络(FFN)组成。它也可以被看作是在全连接几何图上执行消息传递的图神经网络。作者首先解释DGT如何从Gt = (At, xt, ht)中提取信息,然后整合来自0 = (0,0, 0)的信息。对于第一个模块的初始输入,作者将ht和At投影到节点初始特征H1∈ℝN*b1和边缘初始特征E1∈ℝN*N*b2,而P1 ← xt∈ℝN*3是节点的初始三维坐标。对应扩散过程中的时间步长的噪声水平log(αt2/σt2)通过可学习的正弦位置嵌入作为条件特征C∈ℝb3进行投影。
C. 几何图生成过程
使用优化后的数据预测模型dθ,作者通过参数化的逆时间SDE(4)构建了生成性扩散过程。可以使用多种方法从SDE中生成分子,例如Euler–Maruyama方法和祖先采样法。

如图2和算法2所示,作者采用方便的祖先采样方法,并结合数据预测模型和自我条件化技术,生成完整的高质量分子。直观上,作者从先验分布中采样噪声随机数据,并迭代地将其转化为真实数据。原子数量N从训练集中计算的分类分布p(N)中采样,生成过程开始前获取该值。最后,作者对h和A进行简单的取最大值或舍入操作,以获得分子的最终类别或整数变量。
D. 模型变体
作者的扩散模型可以轻松扩展用于具有特定属性c的条件生成G ∼p(G|c)。在实际应用中,作者在DGT中添加条件属性的表示和噪声级别嵌入。通过广泛插入的AdaLN(·)和Scale(·),条件信息可以有效地控制分子三部分的生成过程。如文本等其他条件信号也可以类似地集成到作者的灵活模型中,促进语言引导的分子生成和编辑的潜在应用。如果去除3D组件,作者的模型被称为JODO-2D,可以支持分子图生成和其他图任务。
实验结果
生成质量
表 I
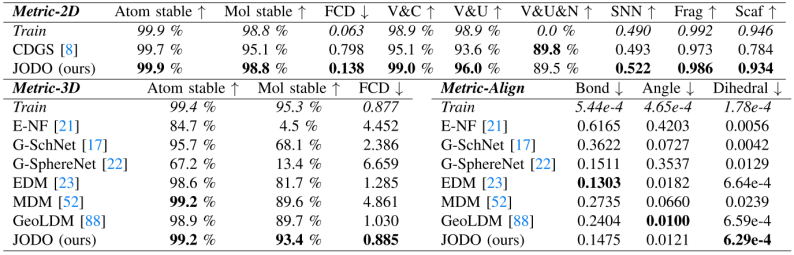
表 II
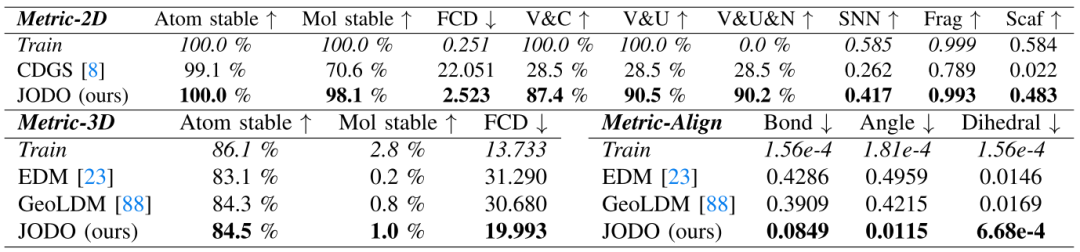
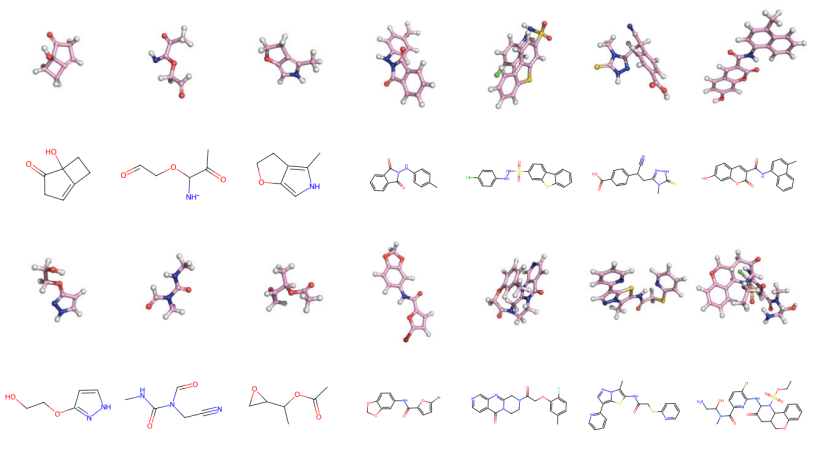
图 3
表 I 和表 II 分别展示了QM9和GEOM-Drugs数据集的结果,图3则展示了一些由该模型生成的分子的可视化。
在 QM9 数据集中,JODO通过2D和3D的联合建模和扩散模型设计,学习精确的几何图分布,生成高度准确的分子。它在2D指标上表现接近最优,并在3D几何指标上显著提高。GEOM-Drugs数据集因每个分子的大量原子而对一次性生成模型提出了巨大挑战。JODO在2D指标,尤其是FCD和V&C指标上大幅超越了先前最先进的基于一次性扩散的CDGS模型,表明JODO能够生成更多有效且连接的药物大小的分子。虽然GEOM-Drugs数据集上的3D稳定性指标可能受到操纵,但更好的模型仍然倾向于具有较低的FCD值和更接近训练集的稳定性比率。因此,作者的模型生成的构象比EDM更为合理。
对于两个数据集上键连接分子图与3D几何之间的对齐,JODO表现出优秀的MMD性能,与测试集中常见子结构几何的分布一致。一个显著的观察是,即使对于训练集中的样本,从3D坐标构建的分子的FCD值也比从拓扑分子描述符计算的低得多,这暗示了化学空间中的不利分布偏移。因此,之前使用的基于规则的高效键后处理方法难以处理更大的分子,这也促使作者开发端到端的联合2D和3D生成模型,直接生成完整的分子。
进一步分析
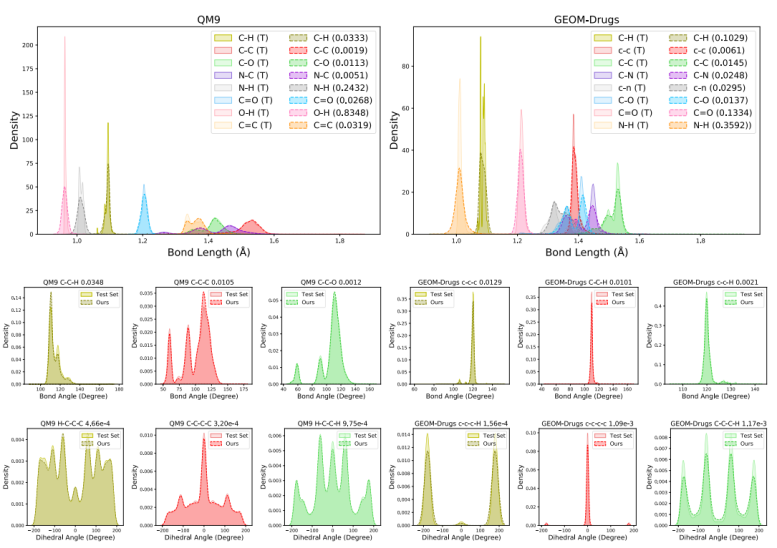
图 4
作者进一步分析了模型在生成对齐良好的2D拓扑和3D几何方面的表现,图4比较了测试集和生成样本中最常见的键长、键角和二面角的分布,而不仅仅报告平均MMD值。可以观察到,不同类型的键长在距离分布上有所不同,而作者的模型很好地捕捉了这种分布差异并拟合了键长的分布形状。性能差距主要来自于一些分布形状具有高峰值的部分,其中键长在较小范围内更加稳定。
对于键角和二面角,即使是那些具有多个峰值的分布,作者的模型在表示其分布方面表现出色。对齐性能表明作者的模型总体上生成了稳定的局部几何。尽管与之前的方法相比,JODO的分布建模能力有所提高,但在某些子结构的几何形状上仍存在一些不足,如GEOM-Drugs数据集中C–C–C的键角、C–N–C–H和C–N–C–C的二面角。这些不对齐现象表明,一些生成的分子的构象可能不处于其基态能量上,未来的工作需要进一步改进。
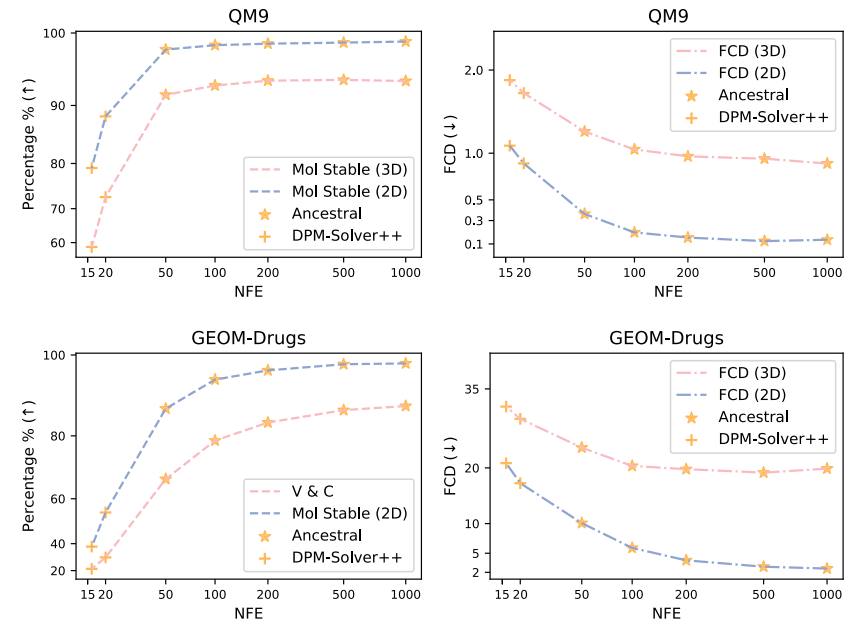
图 5
基于扩散的模型可以通过选择采样中的迭代步数在样本质量和计算成本之间进行权衡。这允许快速采样,有助于通过生成更多分子在合理时间内进行虚拟筛选等应用。图5展示了作者模型的样本质量在不同NFE下的变化。通过使用祖先采样,作者的模型在50到1000 NFE之间生成高保真分子。CDGS成功使用ODE求解器进行快速分子图采样。由于作者的模型在数据空间中保持连续性,作者利用混合采样方法探索了少步数生成(15到20 NFE),使用DPM-Solvers++进行2D图采样,使用祖先采样进行3D坐标采样。
结论
作者提出了一种基于 DGT 的联合 2D 和 3D 图扩散模型,能够生成完整分子的几何图。该模型利用互补的分子描述符来捕捉精确的分子分布,在无条件生成中达到了最先进的性能,并在量子性质的条件生成中显示出显著改善。该模型有潜力应用于药物发现,包括生成大规模的分子库用于结构基础虚拟筛选、逆向设计探索候选分子,以及结合靶点的结构信息生成具有合理化学性质的分子。然而,模型在计算效率和生成复杂分子的质量方面仍存在挑战,未来将扩展到更具挑战性的场景并提高采样效率。
编译 | 于洲
审稿 | 曾全晨
参考资料
Huang H, Sun L, Du B, et al. Learning Joint 2-D and 3-D Graph Diffusion Models for Complete Molecule Generation[J]. IEEE Transactions on Neural Networks and Learning Systems, 2024.
内容中包含的图片若涉及版权问题,请及时与我们联系删除

评论
沙发等你来抢