
DRUGAI
构建准确的蛋白质-配体结合姿态是基于结构的结合亲和力预测的先决条件,这为小分子药物设计中的先导优化提供了结构基础。然而,由于小分子化学空间的复杂性和多样性,对不同分子的结合姿态进行合理的预测是一项挑战。本文介绍的是近期发表在《Journal of Chemical Information and Modeling》上的一篇题为《Fully Flexible Molecular Alignment Enables Accurate Ligand Structure Modeling》的文章。在该论文中,作者开发了一种基于相似性的分子对齐算法Z-align,该算法利用分子的拓扑结构信息作为相似性评价标准,采用了全柔性的分子优化策略,从而实现精确的配体分子结构建模。该论文的第一作者为山东大学物理学院在读博士王志豪,共同通讯作者是郑良振博士(智峪生科)、李伟峰教授(山东大学物理学院)、彭向达博士(智峪生科)。

背景
在小分子药物设计领域,准确预测蛋白质与配体的结合姿态是至关重要的。这种预测不仅为基于结构的结合亲和力预测提供了前提,还为后续的先导化合物优化提供了指导。在酶设计和酶发现中,小分子建模扮演着至关重要的角色,极大地促进了我们对酶的催化机制、底物特异性以及抑制剂设计的理解。预测小分子与酶活性位点的结合模式对于改造酶以适应新的底物或提高催化效率具有指导作用。通过识别关键的结合位点,可以更有针对性地进行突变和重组,以优化酶的催化效率。
然而,由于小分子的化学空间复杂且多样化,合理预测不同分子的结合姿态一直是一个挑战。分子对齐技术是一种基于模板的分子建模方法,在AlphaFold2出现之前,模板建模方法在蛋白质结构预测中展现出了巨大的优势。在蛋白质-小分子复合物的结构预测中,分子对齐方法也同样具有较高的准确性。传统的分子对齐技术,如基于形状的刚性对齐或基于原子级别的柔性对齐,虽然在某些情况下有效,但它们往往依赖于分子间的高相似性。在实际应用中,我们并不能总是找到高相似性的分子,一旦相似性降低,分子对齐算法就难以找到匹配的等效原子对,因此不能生成准确的分子结构。此外,这些方法在处理大分子时,键长和键角的误差会被放大,从而降低对齐的精度。因此,开发一种能够适应不同分子特性、提高对齐精度的新方法,对于推动小分子-蛋白质复合体预测方法的发展具有重要意义。
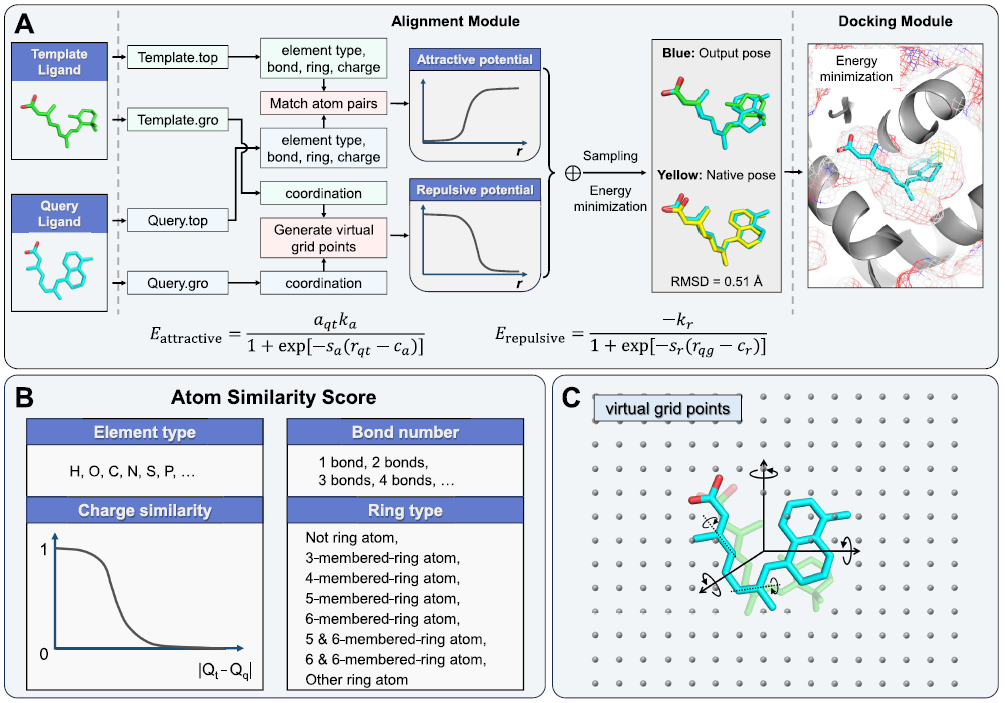
图 1. Z-align算法流程图
研究方法
为了克服现有分子对齐技术的局限性,作者们开发了一种创新的算法——Z-align (Zelixir’s Ligand Alignment Method)。Z-align算法采用了一种全新的对齐方法,它不是依赖于传统的分子指纹相似性,而是利用拓扑结构信息和化学信息来评估查询分子(待优化分子)和模板分子之间原子对的相似性。这里作者采用的等效原子对评估指标包括元素类型、原子电荷、成键数量和成环类型,总的相似性为这几种指标打分的加和:
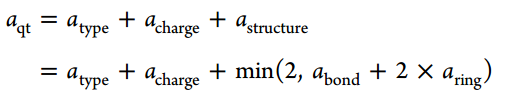
在评估完等效原子对后,算法会为相似性打分大于0的原子对施加吸引势,促使相似原子对的位置尽可能重叠,以对这种构型进行奖励。吸引势能随着原子间距离的增加而迅速减小,这样可以确保相似的原子对在空间上尽可能接近。此外,算法会以模板分子为中心构建虚拟空间格点,以划定查询分子在三维空间中的分布位置。由于格点是根据模板分子的位置生成的,因此难免会与查询分子出现位置重叠碰撞。于是作者为虚拟格点和查询分子之间添加了排斥势能,当查询分子的原子与这些网格点距离过近时,排斥势能会推动查询分子的原子远离网格点,从而避免原子间的重叠。排斥势能具有比吸引势能更长的作用距离,这有助于构建一个平滑的势能表面,减少局部最小值,使分子对齐过程中能够避免陷入局部最优解。
最后,算法会对不同的结构进行采样,并在分子力场的限制下进行能量最小化。这里的总能量包括力场能量以及添加的吸引势能和排斥势能。对于分子对齐任务,算法将会将能量最小的构型输出;而对于分子对接任务,算法会在对齐结果的基础上进一步考虑蛋白跟小分子的相互作用能,再进行能量最小化,输出最佳构型。在该优化过程中,分子的结构受限于力场作用,其内部的键长、键角均可以在能量的作用下微调,因此可以减小优化过程中的误差积累,是一种全柔性分子对齐方法。
研究结果
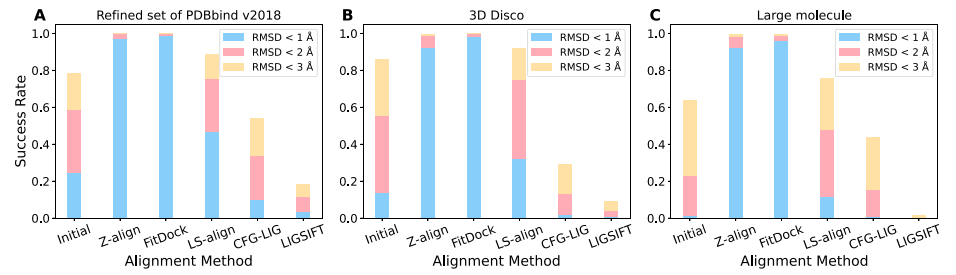
图 2. 不同算法的自对齐性能对比
图2展示了Z-align算法与其他四种分子对齐方法(FitDock、LS-align、CFG-LIG和LIGSIFT)在自对齐任务中的性能对比。作者使用PDBbind v2018和3D Disco数据集,通过计算随机构象与原生构象之间的RMSD值来评估对齐的准确性。Z-align表现出色,在2 Å RMSD阈值下,对PDBbind和3D Disco数据集的配体自对齐成功率分别达到了99.73%和98.63%。在大分子数据集上仍能保持较高的成功率,说明Z-align算法可以有效减少大分子中结构误差的积累。
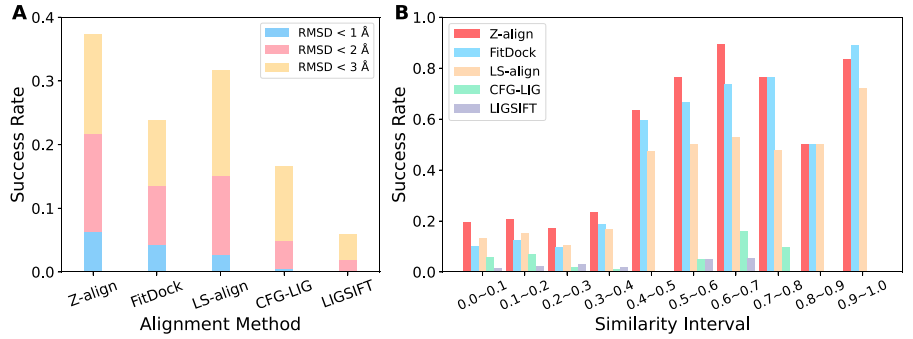
图 3. 不同算法的模板对齐性能对比
模板对齐是用已知结合口袋中的配体作为模板,将其他小分子的随机构象对齐到这个模板上。图3A表明,在3D Disco数据集上,Z-align在模板对齐任务中的成功率最高。图3B进一步分析了在不同相似性区间的模板对齐成功率,揭示了Z-align在中等相似性区间(0.4到0.8)的特别优势,这表明Z-align能够有效地识别和利用分子间的拓扑相似性,即使在分子间整体相似性不高的情况下,也能实现有效的结构对齐。在药物设计中,潜在的药物分子需要与模板分子具有一定相似性但又需要存在结构差异以实现特定生物功能,因此这一结果强调了Z-align在实际药物设计和酶设计中的潜在应用价值。
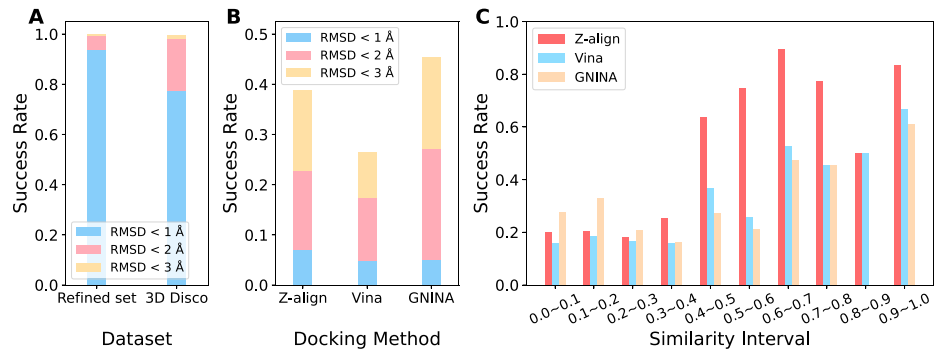
图 4. 对接性能测试
作者也评估了Z-align算法在分子对接性能方面的表现,特别是在redocking和cross-docking测试中。redocking测试中(图4A),Z-align展现出接近完美的成功率,RMSD小于2 Å的成功率接近100%,这表明了Z-align在已知配体结构的情况下预测结合姿态的高准确性。在cross-docking测试中(图4B),作者把Z-align与AutoDock Vina和GNINA算法进行了比较。结果显示,尽管GNINA在低相似性配体对中表现出色,Z-align在高相似性区间(超过0.4)的配体对中的成功率显著提高,表明其在模板基础对接方面的强大能力。图5C进一步细分了不同相似性区间的cross-docking成功率,揭示了Z-align在处理具有一定结构相似性的配体时的优越性能。这些结果强调了Z-align算法在药物设计中的实用价值,尤其是在需要利用已知结构信息来预测分子结合姿态的场景中。
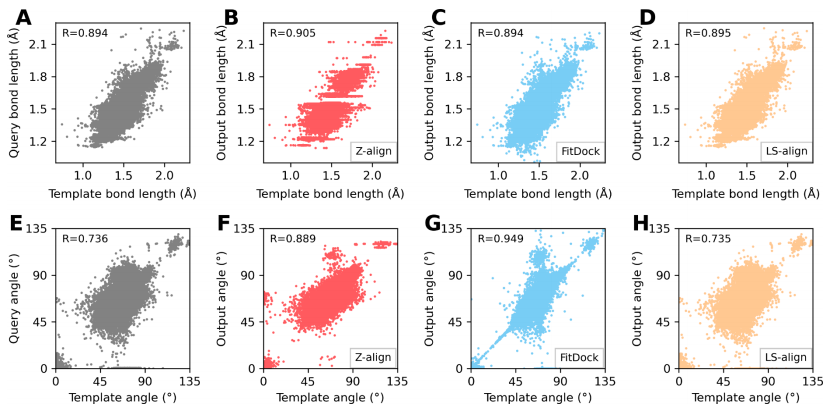
图 5. 算法优化前后分子的键长、键角相关性
图5对Z-align算法在处理大分子数据集时对分子键长和键角优化的能力进行了深入分析。通过比较模板分子与经过不同算法处理后的输出分子之间的键长和键角,揭示了Z-align在保持分子几何准确性方面的优势。
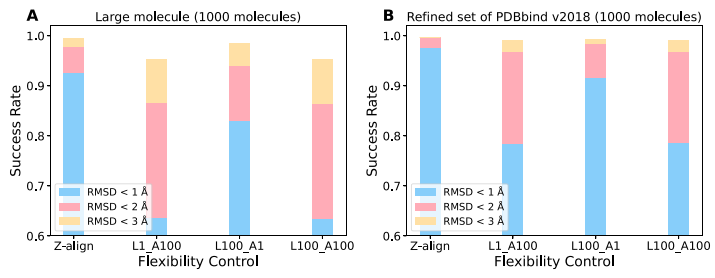
图 6. 不同柔性强度下Z-align算法的成功率
图6探讨了分子对齐过程中键长和键角灵活性对成功率的影响。实验结果表明,增加键长、键角的刚度系数会使成功率降低,而键角刚度系数对成功率的影响更为显著。这些发现说明,在分子对接中保持适当的灵活性是至关重要的,同时也证明了Z-align算法在不同大小的分子中都能保持较高的对齐成功率,这得益于其在优化过程中对分子键长和键角的灵活调整能力。
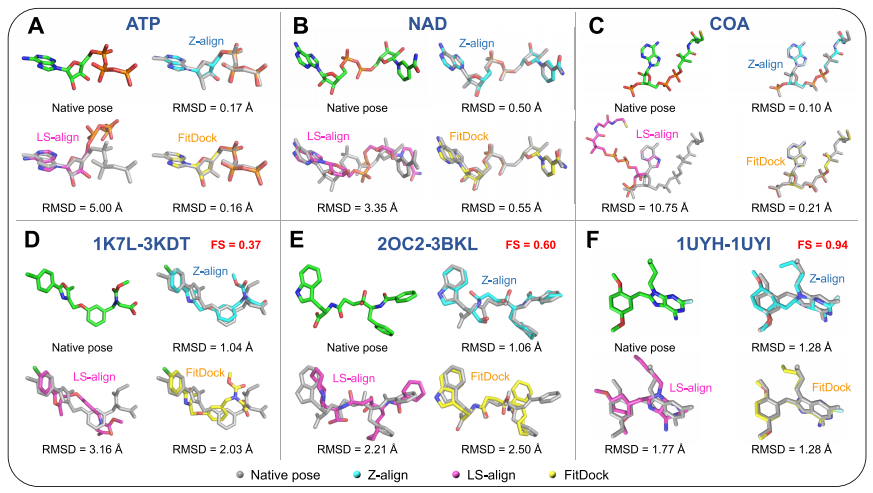
图 7. 不同算法的对齐结果示意图
参考资料
Wang, Z., Zhou, F., Wang, Z., Hu, Q., Li, Y. Q., Wang, S., Wei, Y., Zheng, L., Li, W., & Peng, X. (2024). Fully Flexible Molecular Alignment Enables Accurate Ligand Structure Modeling. Journal of chemical information and modeling, 10.1021/acs.jcim.4c00669.
https://doi.org/10.1021/acs.jcim.4c00669
内容中包含的图片若涉及版权问题,请及时与我们联系删除

评论
沙发等你来抢