
DRUGAI
今天为大家介绍的是来自Mihaela Angelova团队的一篇论文。多重成像规模和维度的增长需要可重复且全面但用户友好的计算流程。TRACERx-PHLEX包含基于深度学习的细胞分割(deepimcyto)、自动化细胞类型注释(TYPEx)和可解释的空间分析(Spatial-PHLEX)这三个独立但可互操作的模块。PHLEX生成单细胞身份、组织区室内的细胞密度、标记阳性调用和诸如细胞屏障评分等空间指标,以及总结图和空间可视化。PHLEX是在TRACERx研究中使用成像质谱细胞术(IMC)开发的,并通过已发表的索引共检测(CODEX)、IMC和正交数据进行验证,并与最先进的方法进行对比。作者评估了其在不同组织类型、组织固定条件、图像大小和抗体面板上的应用。由于PHLEX是一个自动化和容器化的Nextflow流程,手动评估、编程技能或病理学专业知识不是必需的。PHLEX在多重数据领域提供了一个端到端的解决方案,并提供了临床相关的见解。可以通过https://github.com/FrancisCrickInstitute/TRACERx-PHLEX体验PHLEX,可通过https://tracerx-phlex.readthedocs.io/en/main/ 查看详细的文档介绍。

高维组织成像使得原位详细细胞表型分析成为可能,这对于理解细胞功能和协调至关重要。随着同时分析的标记和样本数量的增加,数据分析和解释的复杂性也在增加。多重成像分析通常包括五个基本步骤:图像预处理、细胞分割、细胞表型分析、空间分析和可视化。
最近,独立的概率方法使得使用多重成像的细胞亚型的自动识别和注释成为可能,前提是用户提供细胞亚型特异性标记的列表和分配概率的阈值。虽然深度学习方法不限于预先定义的亚型,但可能需要针对特定抗体面板的训练数据。因此,需要能够全面识别细胞亚型和功能标记阳性的全自动方法。
为了解决这些需求,作者开发了TRACERx-PHLEX(多重成像的细胞表型和定位分析),这是一个综合且用户友好的多重成像分析和解释流程。作为一个模块化的Nextflow流程,PHLEX首先结合了一个基于深度学习的分割模型和用于IMC的真实数据集。其次,它通过细胞亚型识别、分配和标记阳性调用,自动完成复杂的细胞表型分析任务。第三,PHLEX结合了一套空间分析方法,便于测量临床相关的组织结构特征,例如肿瘤细胞被基质细胞广泛封闭。
系统工作流程
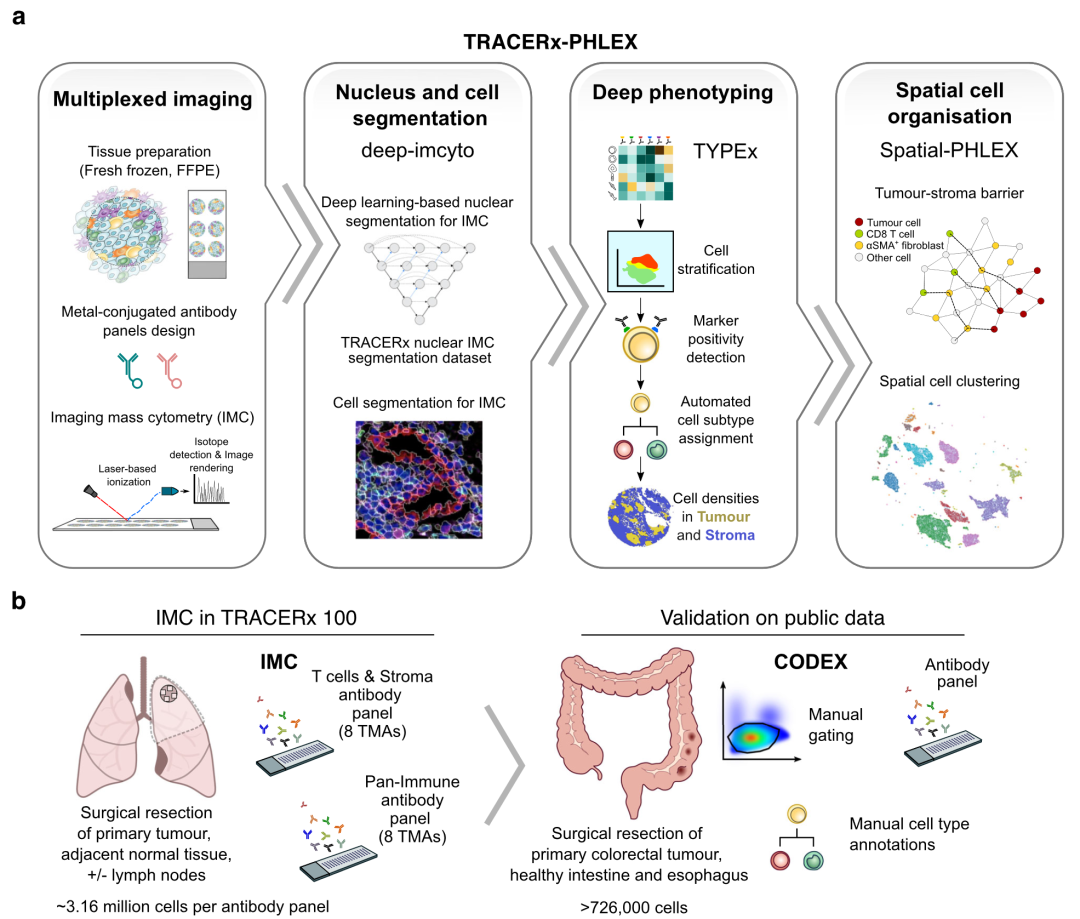
图 1
这里,作者介绍了PHLEX,这是一种用于多重成像的细胞表型和定位分析的自动化工作流程(图1a)。为了解决IMC数据分析中的关键挑战,作者开发并集成了PHLEX工作流程中的三个模块:(i)deep-imcyto用于细胞核和全细胞分割,(ii)TYPEx用于深入识别细胞亚型和细胞状态,以及(iii)Spatial-PHLEX用于可解释的空间分析。这些模块是可互操作的,并作为独立流程封装在Nextflow中,Nextflow是一个管理工作流程的平台,具有所有软件依赖项的容器化功能。因此,PHLEX的模块也可以作为独立工具使用。TYPEx和Spatial-PHLEX模块可以更广泛地应用于IMC以外的多重成像数据。
作者展示了PHLEX在通过治疗跟踪癌症演变(TRACERx)100队列研究中的应用(ClinicalTrials.gov标识符:NCT01888601),使用IMC对83名非小细胞肺癌(NSCLC)患者的236个TMA核心进行了分析。此外,作者评估并将其性能与标准工具进行了对比,使用了公开的包括人类和小鼠数据、IMC和索引共检测(CODEX)平台、小型TMA和大型整片图像,以及不同的组织类型、组织固定条件和抗体面板(图1b)。作者分析了来自肿瘤及其邻近肺部组织、结直肠癌、乳腺癌、健康肠道、巴雷特食管和淋巴组织的成像数据。
deep-imcyto:用于IMC的细胞核和全细胞分割
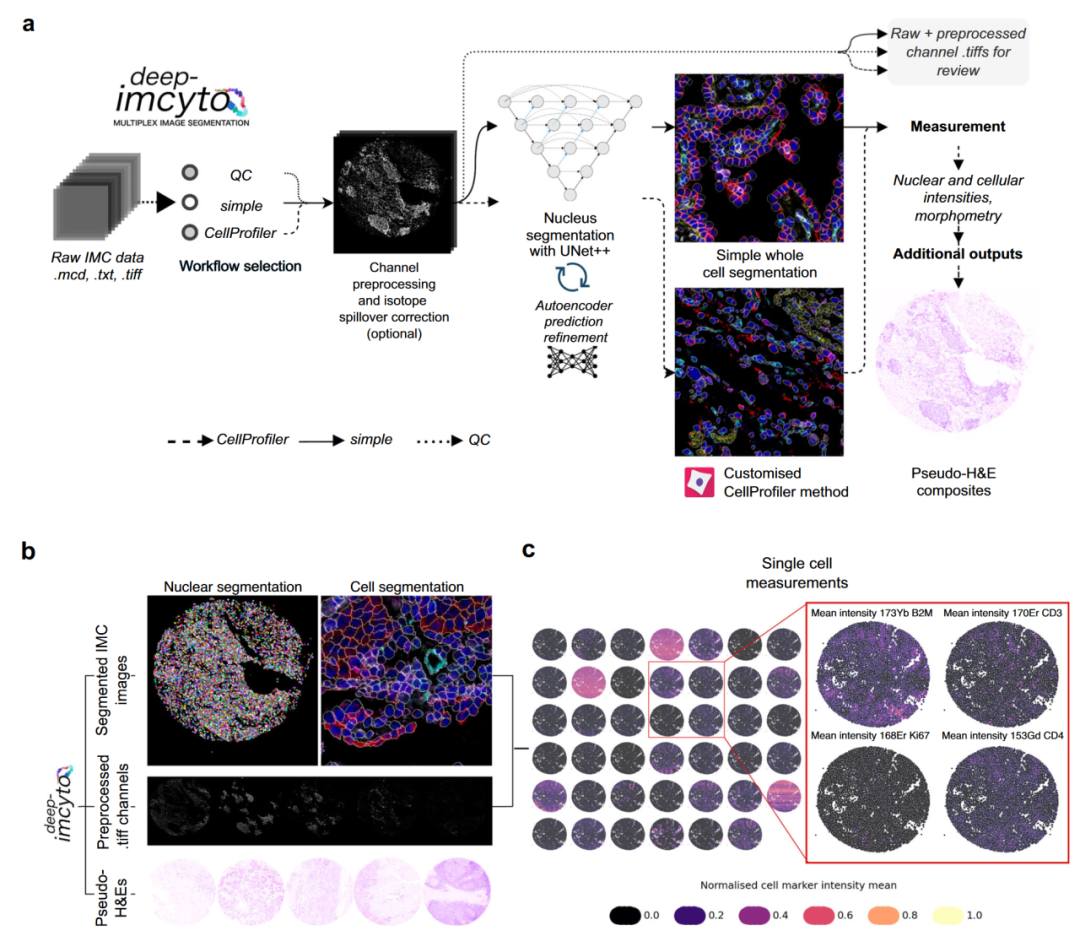
图 2
作者之前描述了nf-core/imcyto IMC分割流程的开发。在此基础上,作者推出了deep-imcyto(图2a),这是一个灵活的、自动化的基于深度学习的Nextflow流程,用于IMC图像的细胞分割,具有多种可自定义的工作流程选项。这些选项包括数据质量控制和两种细胞分割流程,用户可以根据应用需求进行选择。deep-imcyto模块满足了在细胞表型分析和空间分析之前,自动准确定义细胞边界和测量细胞标记物强度的需求。
deep-imcyto使用高度准确的细胞核分割作为IMC图像中全细胞分割的基础,通过使用L4 UNet++模型架构的实例级细胞核分割过程实现这一点,该模型在内部真实数据集上训练,并结合基于深度自编码器的异常检测。为了解决模型训练所需的注释IMC数据集需求,两位专家手动标注了来自TRACERx 100 IMC组织核心的41,962个形态各异的细胞核,涵盖了多种肺癌组织亚型和结构,以及组织学上正常的肺、淋巴结、扁桃体和肾组织。作者提供了这一TRACERx细胞核IMC分割数据集,据作者所知,这是目前IMC领域最大的此类数据集,作为研究社区的资源。
在deep-imcyto的细胞核分割步骤之后,作者提供了两种工作流程选项,用户可以基于UNet++细胞核种子来确定全细胞边界:简单模式和CellProfiler模式(图2a)。对于全自动细胞分割程序,deep-imcyto默认的简单分割工作流程通过用户定义的像素数量扩展深度学习模型的细胞核掩码,以近似细胞边界。这种端到端选项从输入图像生成单细胞掩码,无需额外配置。简单工作流程可以快速处理大型输入数据集:作者对Jackson等人的746个感兴趣区域(ROI)进行了处理,每个ROI的平均执行时间为2.0分钟。然而,像素扩展方法可能无法捕捉多样的细胞形态,或者在细胞拥挤区域表现不佳,也无法捕捉到没有平面细胞核的细胞。出于这些原因,deep-imcyto像nf-core/imcyto一样,允许提供一个CellProfiler流程,在识别细胞核之后执行用户定义的细胞分割步骤。作者使用deep-imcyto的CellProfiler模式进行定制分割,利用IMC的多通道信息来改进细长基质细胞的识别,旨在更好地区分肺肿瘤微环境中各种细胞类型的形态,这种方案被称为多重共识细胞分割(MCCS)。
从简单模式或CellProfiler模式生成的全细胞掩码用于提取单细胞级别的标记物强度、细胞-细胞邻近数据和形态特征。作为一个附加功能,deep-imcyto从输入的IMC文件生成伪苏木精-伊红(H&E)图像,无需使用钌对比染色,以便直接进行病理学家的IMC图像标注(图2b)。单细胞强度数据可以以细胞-标记物矩阵的格式输入到细胞表型分析模块TYPEx,用于识别细胞表型和细胞标记物阳性状态(图2c)。
TYPEx在组织微环境中深入识别细胞表型
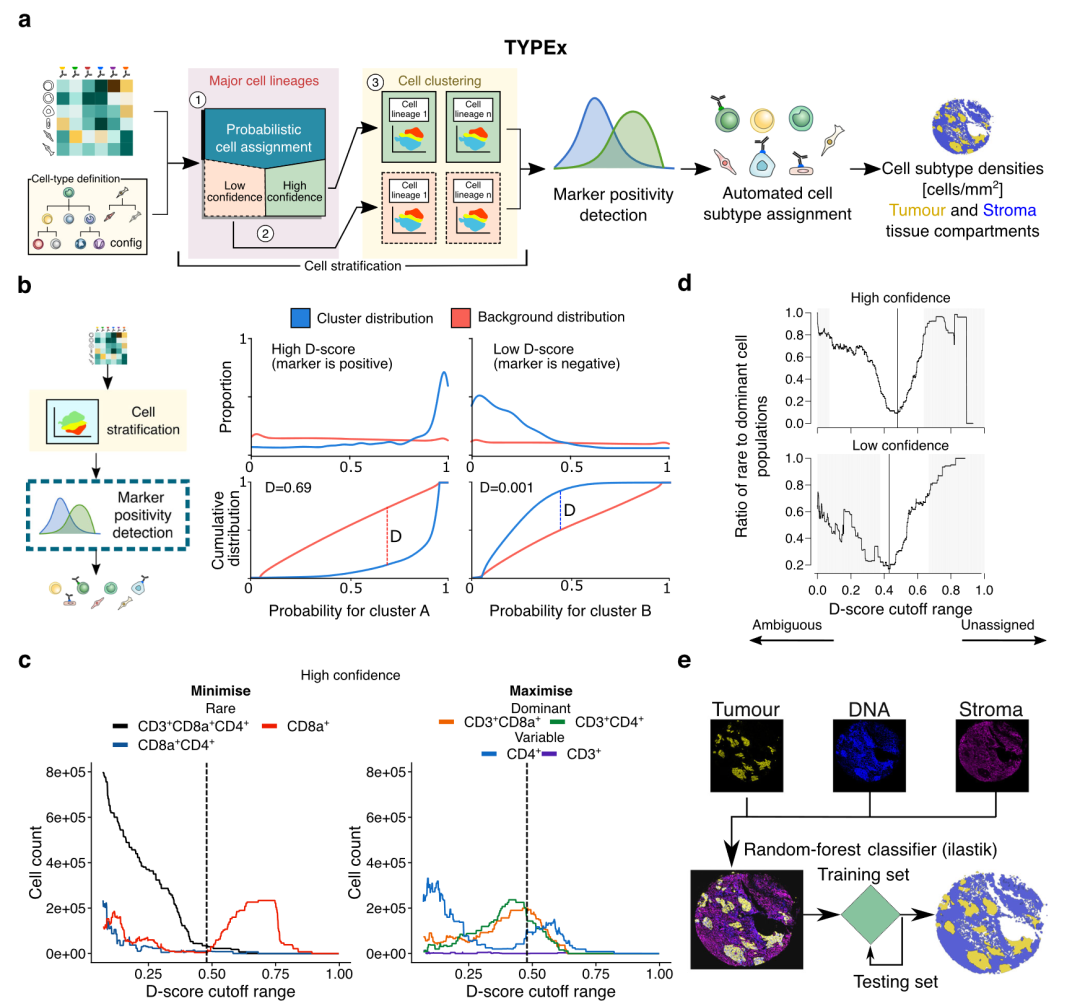
图 3
细胞表型分析模块TYPEx满足了自动识别和分配细胞类型以及全面检测单个分割细胞状态的需求。为此,TYPEx检测标记物的阳性情况,解决低置信度注释问题,并基于细胞上的阳性标记物组合分配细胞亚型(图3a)。所需的输入是一个细胞-标记物强度矩阵和一个包含用户抗体面板定制的细胞类型定义的文件,这种格式允许灵活应用于其他成像模式。TYPEx首先基于现有工具进行细胞分层,然后采用统计方法确定标记物阳性情况。基于阳性标记物的组合,它会根据用户提供的细胞类型定义自动注释细胞亚型。这里,作者详细介绍了TYPEx的四个分析步骤:细胞分层、标记物阳性检测、细胞亚型分配和组织分割,并以TRACERx 100 IMC数据集为例进行展示。
细胞分层步骤旨在将细胞根据类似的标记物强度水平进行分组,并根据主要细胞谱系标记物的强度区分低置信度和高置信度组。低置信度细胞可能位于密集的免疫区域,如淋巴细胞聚集区,信号可能会溢出到邻近细胞。此外,低置信度细胞可能具有较弱的强度和低信噪比的细胞谱系特异性标记物。TYPEx中的三层细胞分层方法结合了现有的概率和无监督方法,首先按假定的主要细胞谱系分层,其次按置信度分层,最后按细胞聚类分层(图3a)。
为了避免在检测标记物阳性时设置全局信号强度阈值,将给定聚类中的标记物强度转换为概率,即聚类中的一个随机细胞比另一个聚类中的随机细胞具有更高强度的给定标记物的概率。通过D评分来总结给定聚类的概率分布与背景分布的偏差,D评分表示累积概率分布之间面积的差异(图3b)。每个测量的标记物都会分配一个D评分给每个聚类。表达给定标记物的聚类将具有较高的D评分,反映出其概率分布相比背景分布偏向较高值。
TYPEx计算出一个最佳的D评分阈值,并在给定聚类的D评分高于该阈值时,判定该聚类的标记物为阳性。最佳阈值是基于已知的一部分标记物的共表达模式来确定的。默认情况下,TYPEx使用T细胞标记物CD4、CD8a和CD3,例如,在外周非淋巴组织中,双阳性()和单阳性细胞预计很少见,而和细胞预计是分析队列中的主要群体(图3c)。基于这些标准,TYPEx通过最小化稀有T细胞群体与主要T细胞群体的比例来估算最佳D评分阈值,尽量减少稀有群体并最大化主要群体。如果高估了D评分阈值,会导致单阳性CD8a+细胞比例更高和未分配的调用增多;如果低估了D评分阈值,会导致双阳性CD4+CD8a+ T细胞(CD3+/-)比例更高和模糊调用增多(图3d)。
为了量化每个组织区域内以及肿瘤和基质组织区室内的细胞注释,作者使用ilastik在来自肿瘤、相邻正常肺组织和淋巴结组织的图像上训练了一个三分类随机森林模型,用于背景、肿瘤巢/上皮和基质区域分割(图3e)。为了在不同组织类型中更广泛应用,TYPEx还包含一个用于组织和背景分割的两分类模型。此外,它还接受用户提供的二值掩码作为输入,例如病理注释,并标注这些区域内的细胞对象。细胞表型分析的时间和计算复杂度可能会根据细胞数量、测量的标记物数量和数据集中的主要细胞谱系数量而有所不同。
TYPEx输出各种摘要表格、图表、像素级信息的可视化以及注释单细胞对象的地图,以便进行数据查询、解释和细胞表型结果的质量控制(图4)。输出的细胞对象表格可以直接用作Spatial-PHLEX的输入。
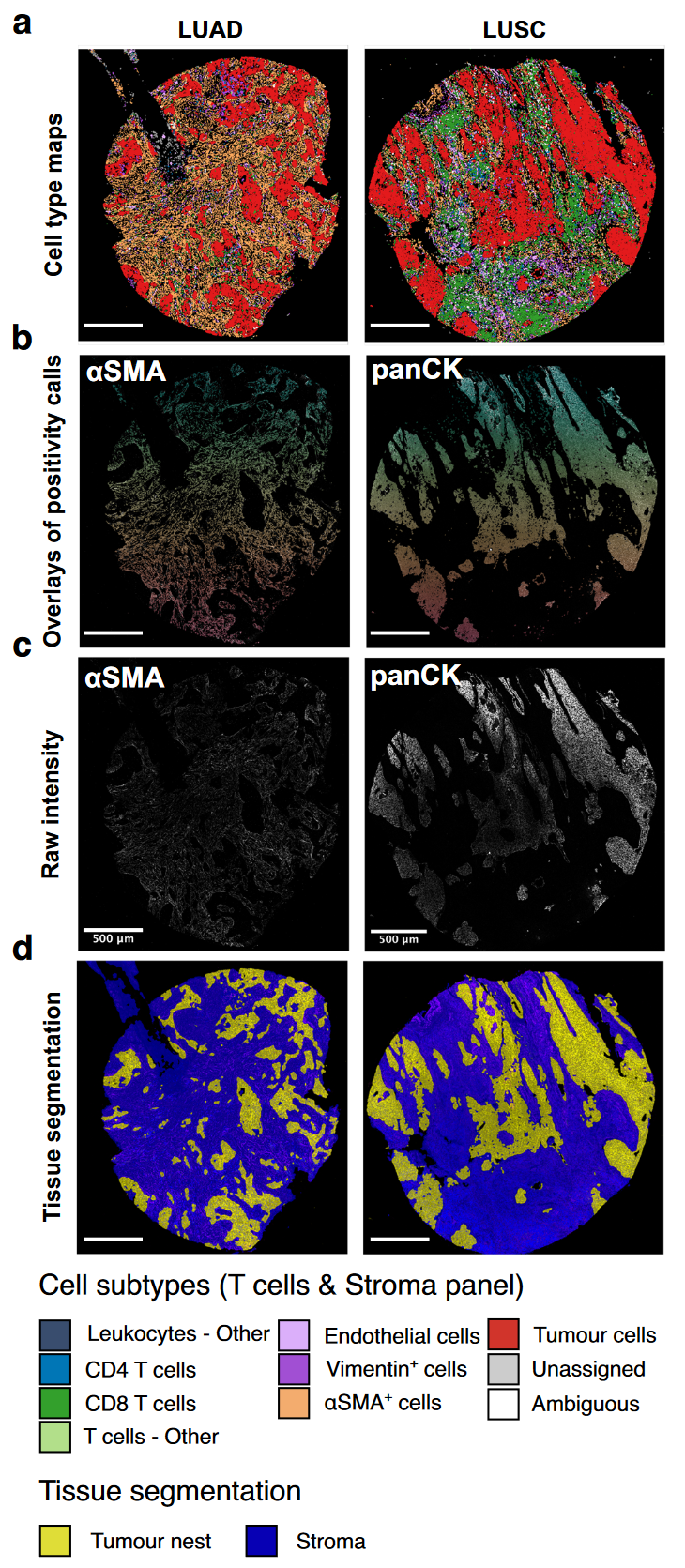
图 4
编译 | 黄海涛
审稿 | 曾全晨
参考资料
Magness, A., Colliver, E., Enfield, K. S., Lee, C., Shimato, M., Daly, E., ... & Angelova, M. (2024). Deep cell phenotyping and spatial analysis of multiplexed imaging with TRACERx-PHLEX. Nature Communications, 15(1), 5135.
内容中包含的图片若涉及版权问题,请及时与我们联系删除

评论
沙发等你来抢