
DRUGAI
2024年8月27日,清华大学生命科学学院/北京生物结构前沿研究中心龚海鹏课题组在《Nature Communications》杂志发表了题为“An end-to-end framework for the prediction of protein structure and fitness from single sequence”的文章。该研究提出了单序列蛋白质结构预测模型SPIRED,在 CAMEO /CASP15 测试集上与 OmegaFold 精度相当,在 SCOPe结构分类数据库上的预测精度超过了ESMFold。并且,SPIRED的推理速度大约是它们的 5 倍,训练时间消耗仅为它们的十分之一。通过将SPIRED与图神经网络集成为一个端到端的网络框架SPIRED-Fitness,能够快速预测蛋白质适应度以及结构。在ProteinGym数据集的零样本(zero-shot)与有监督(supervised)测试方式上,SPIRED-Fitness的预测精度超过了绝大部分的单序列模型,接近最先进的基于MSA的模型。此外,以SPIRED-Fitness的参数为起点训练得到的SPIRED-Stab模型,在预测突变对蛋白质稳定性影响方面达到了最先进的性能。并且,作者提供了SPIRED-Fitness与SPIRED-Stab网页服务器与相关代码供广大科研工作者免费使用。

背景
近年来,研究人员将预训练的蛋白质语言模型与结构预测网络结合,构建了以ESMFold2、OmegaFold为代表的单序列蛋白质结构预测模型,实现了快速且较高精度的结构预测,速度比AlphaFold2提升了数十倍。但是,由于这些单序列模型的结构预测网络大多参考AlphaFold2的网络架构,因此它们存在训练代价大、推理时间依然较长、难以与下游功能预测模型进行端到端训练等问题。另一方面,目前大部分有监督的蛋白质适应度预测模型需要在每一个蛋白质上重新训练一个模型,且推理速度较慢,因而极大地增加了模型训练与应用成本。
模型亮点
SPIRED 采用了创新的模型设计,即顺序排列的折叠单元(Folding Units),并使用了相对位移损失(Relative Displacement Loss, RD Loss)以显著提高计算效率。与ESMFold不同的是,每个Folding Unit都预测结构坐标,并接受RD Loss约束。并且,与FAPE Loss相比,RD Loss更关注残基间相对位移的预测值与真实值的差距。并且,RD Loss 省去了FAPE Loss计算过程中所需要的旋转矩阵预测以及坐标对齐,只需要SPIRED预测局部坐标系内的残基间相对位置,从而减小了模型训练的难度。
该研究将 SPIRED 与图神经网络集成为一个端到端的网络框架SPIRED-Fitness,利用Soft Spearman Loss作为损失函数使SPIRED-Fitness在一次训练中学习485种蛋白质的超过65万突变数据。该研究沿用先前工作GeoFitness的思路,将不同类型的蛋白质适应度数据(酶活性、亲和性、稳定性等)转化为突变效应的排序数据,避免不同类型数据的数值尺度与量纲不同带来的训练问题,并且利用Soft Spearman Loss使得排序数据在模型学习过程中可导。
以SPIRED-Fitness 的网络参数为起点,在∆∆𝐺和∆𝑇𝑚数据集上继续训练得到SPIRED-Stab模型。SPIRED-Stab模型的输入为野生型序列和突变序列,能够预测任意突变导致的蛋白质稳定性变化。由于野生型序列和突变序列共享绝大部分模型参数,因此确保了正向突变与反向突变的∆∆𝐺和∆𝑇𝑚预测值的反对称性。
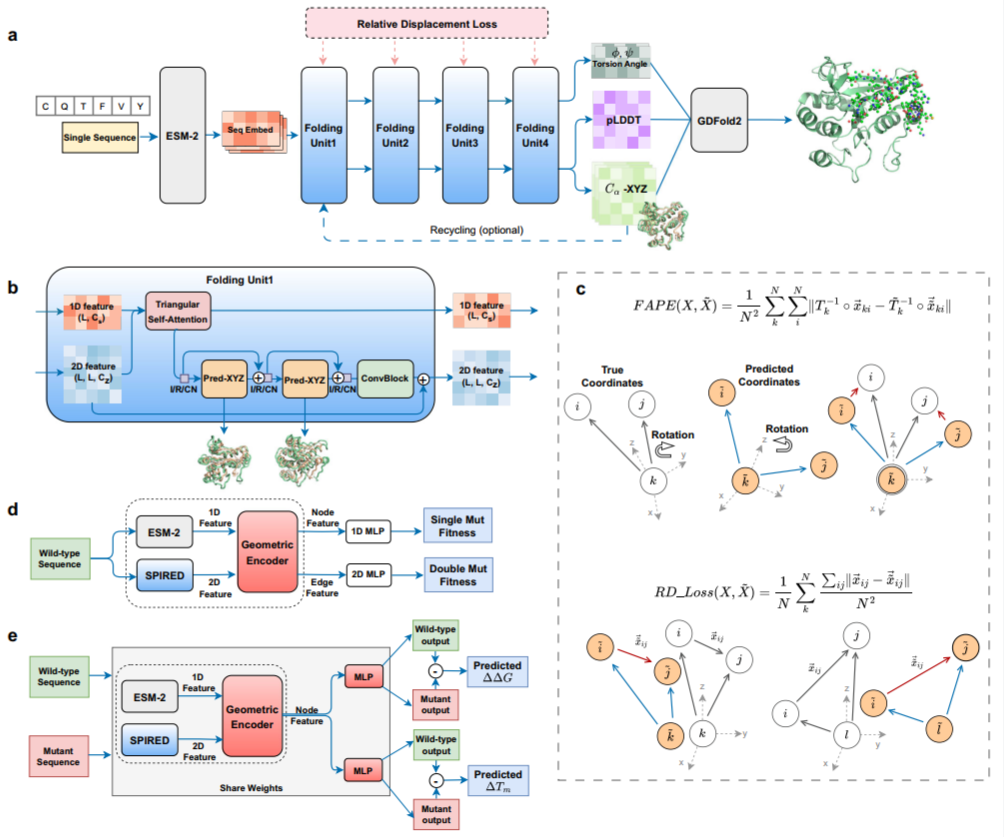
图1. 网络架构与损失函数. a. SPIRED的网络架构;b. Folding Unit的内部网络结构;c. RD Loss与FAPE Loss的差异;d. SPIRED-Fitness的网络架构;e. SPIRED-Stab的网络架构。
主要实验结果
在Cycle=1的情况下,SPIRED-Fitness在CAEMO与CASP15测试集上都超过了单序列版本的AlphaFold2,并且与OmegaFold的平均TM-score几乎持平。在蛋白质结构分类数据库SCOPe上测试,SPIRED 在1231种蛋白质折叠类型(Fold)的平均TM-score上达到0.794,高于ESMFold(0.777)与OmegaFold(0.748),其中91种折叠类型的TM-score值高于ESMFold并且差值超过0.2。另外,SPIRED的推理速度是ESMFold与OmegaFold的4~5倍。在众多蛋白质折叠类型上的展现出的均衡的预测精度以及快速的推理速度,体现出SPIRED在蛋白质功能预测与蛋白质设计上的应用潜力。
其中值得关注的是,SPIRED 在绿色荧光蛋白(GFP)结构预测上显示出了优势。GFP 是蛋白质工程与蛋白质设计中备受关注的蛋白之一。SPIRED在GFP这一折叠类型(SCOPe Fold ID: d.22)中所有蛋白质的平均TM-score为0.959,而ESMFold 与 OmegaFold分别只有0.485与0.577。
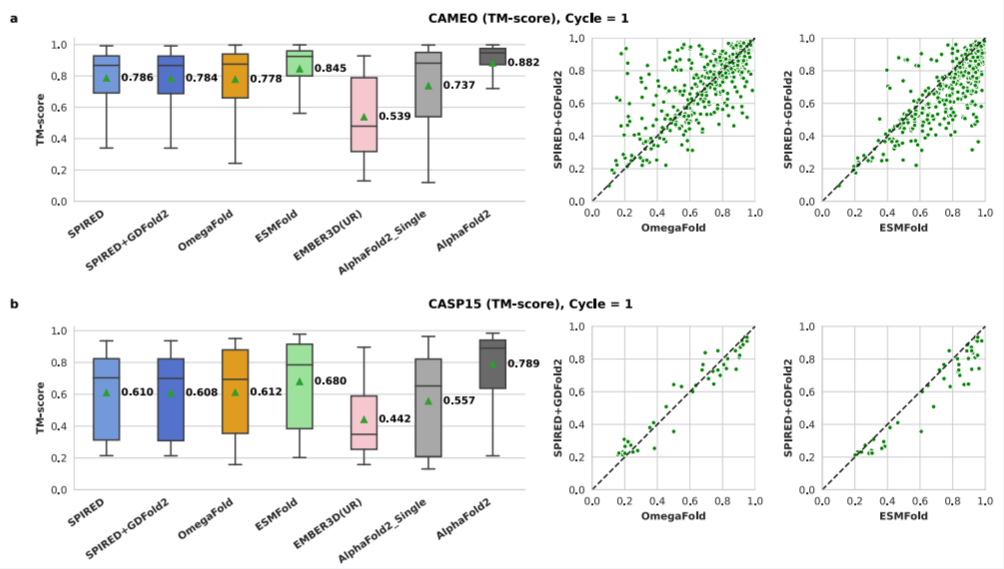
图2. CAMEO与CASP15测试集上的结构预测精度
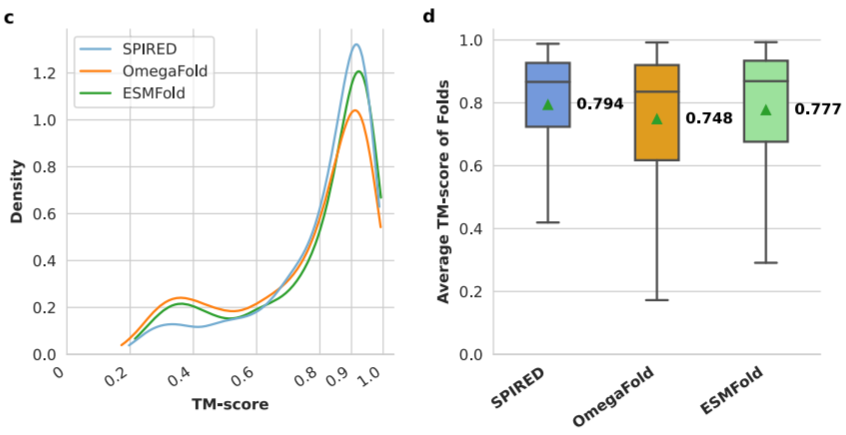
图3. SCOPe(v2.08,S95)结构分类数据库上的预测精度
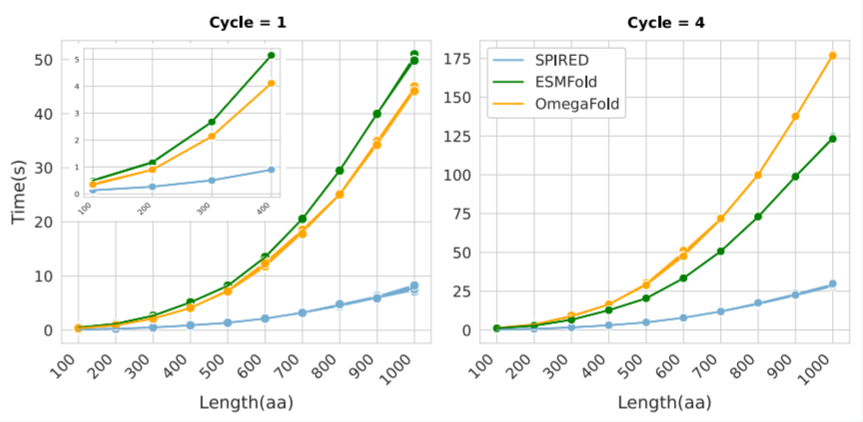
图 4. 模型推理时间的比较
SPIRED-Fitness的训练分为两个阶段:在第一个阶段的训练过程中,SPIRED的参数冻结,只更新图神经网络参数;第二阶段的训练过程中,SPIRED参数解冻。端到端的训练使SPIRED-Fitness的适应度预测精度(Spearman相关性系数)提升了约2%,超过了ECNet。并且,SPIRED的推理速度也远高于传统有监督模型(如ECNet),可快速(秒级别)、精确地预测蛋白质适应度。另外,SPIRED-Fitness模型具有较强泛化性,并且对稀疏训练数据展现出很强的鲁棒性:当训练样本仅有总数据量的10% 时,SPIRED-Fitness在测试集上Spearman 相关系数仍保持在0.7以上,而ECNet的Spearman相关系数下降至0.4以下。
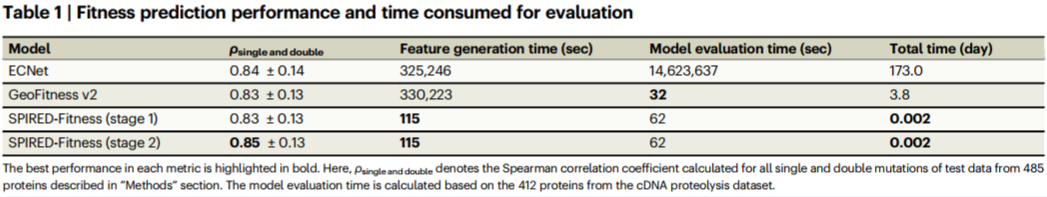
表1. 适应度预测模型的精度与时间消耗
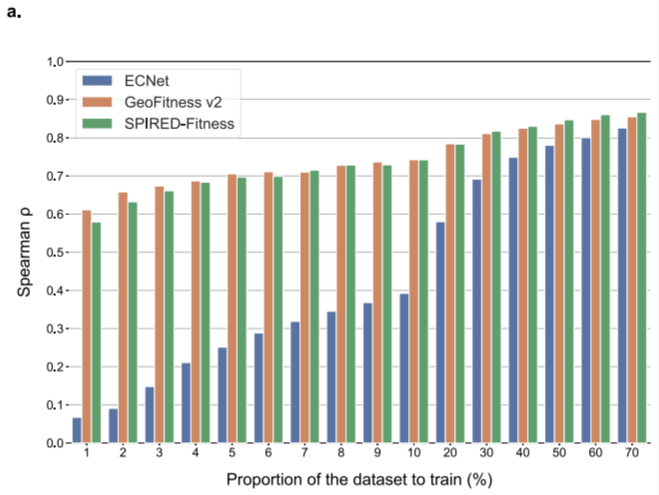
图5. SPIRED-Fitness对稀疏训练数据的鲁棒性
在ProteinGym测试集上SPIRED-Fitness 展现出较高的预测精度水平:(1)在零样本预测(zero-shot)模式下,SPIRED-Fitness 的 Spearman相关性系数(𝜌)、MCC 、AUC指标超过所有参评的基于单序列的模型,很接近基于MSA的TranceptEVE L模型的精度(AUC 与 MCC 相同,单突变的 𝜌 只相差 0.01);(2)在有监督学习模式下,SPIRED-Fitness是精度最高的单序列模型,并超过大部分基于MSA的模型,仅次于ProteinNPT。
在∆∆𝐺测试集S669与S461上,SPIRED-Stab的Spearman 相关系数、Pearson 相关系数、Kendall秩相关系数指标超过了其他所有参评的模型,包括近期出现的优秀模型如RaSP、Pythia、ThermoMPNN、PROSTATA等。在∆𝑇𝑚测试集 S557上,SPIRED-Stab的Spearman相关性系数、Kendall秩相关性系数以及Top-5精度指标也达到最高水准。
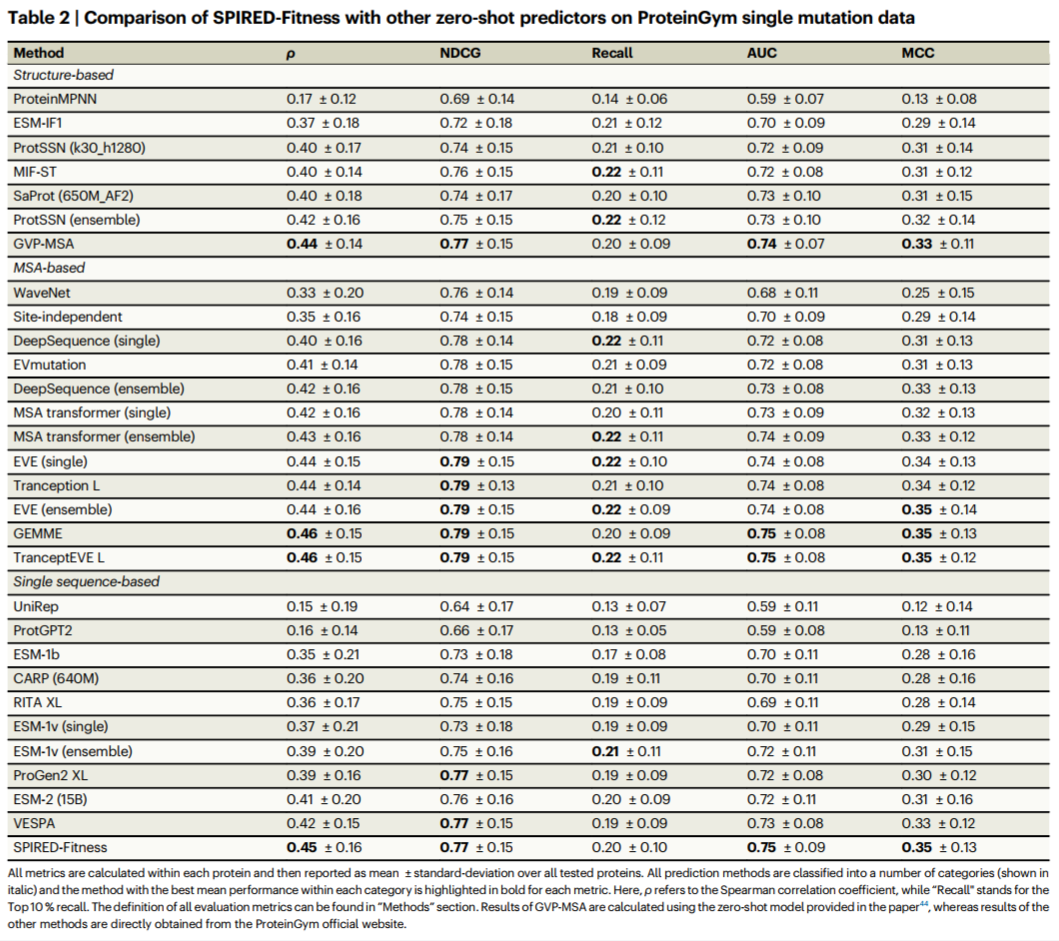
表2. ProteinGym上单突变的zero-shot预测表现
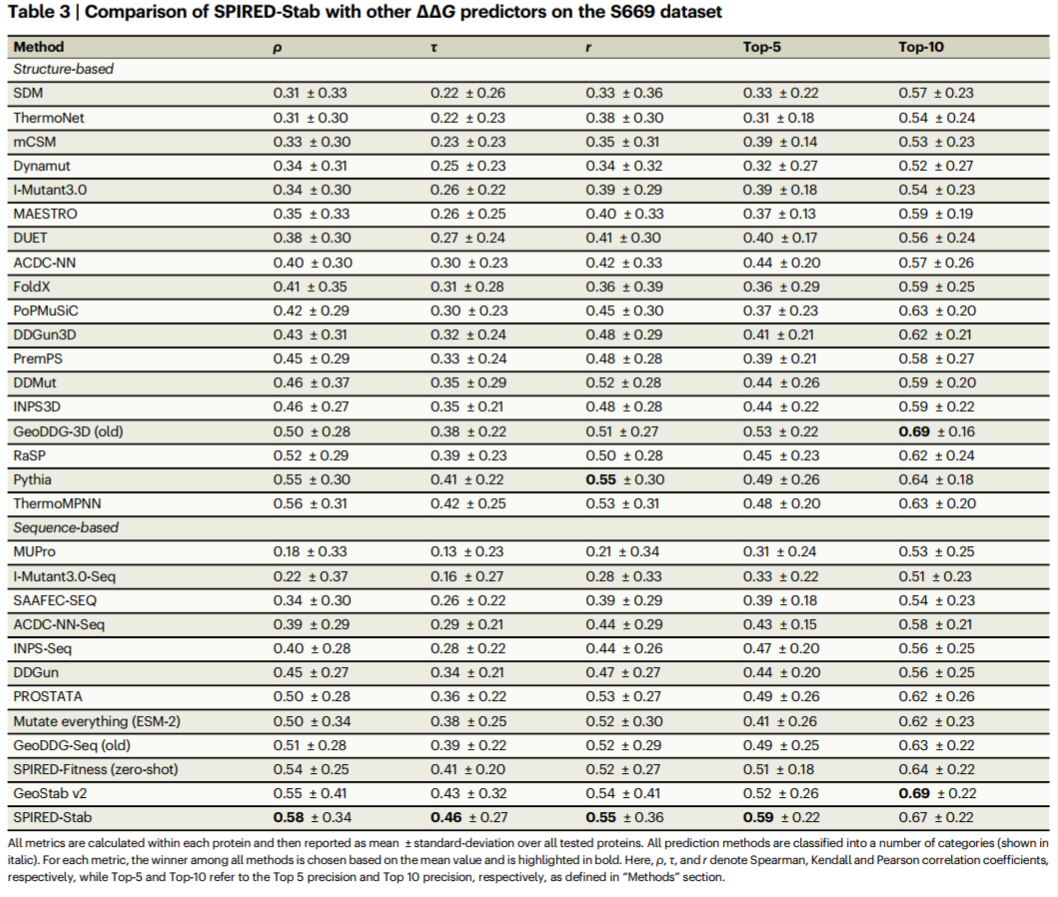
表3. ∆∆𝐺测试集S669上的预测表现
总结与讨论
该研究通过创新性的网络与损失函数设计,构建了低训练消耗、高推理速度、高精度的结构预测模型SPIRED。并且,将SPIRED与图神经网络进行端到端训练提升了SPIRED-Fitness的适应度预测精度。SPIRED-Fitness只需要一次训练即可预测任意蛋白质的适应度与结构,再加上其极快的推理速度与高预测精度,很大程度地提升了适应度预测的效率与便捷性。该研究利用了分层递进的训练方式,依次利用了序列、结构、适应度、稳定性数据,最后得到的SPIRED-Stab模型实现了目前最高的蛋白质稳定性变化预测精度水准。由于生物学、医学数据普遍存在“多种不同类型标签”、“碎片化”的特点,因此该研究中的预训练方式也可推广到其他生物学特性预测模型的训练当中。
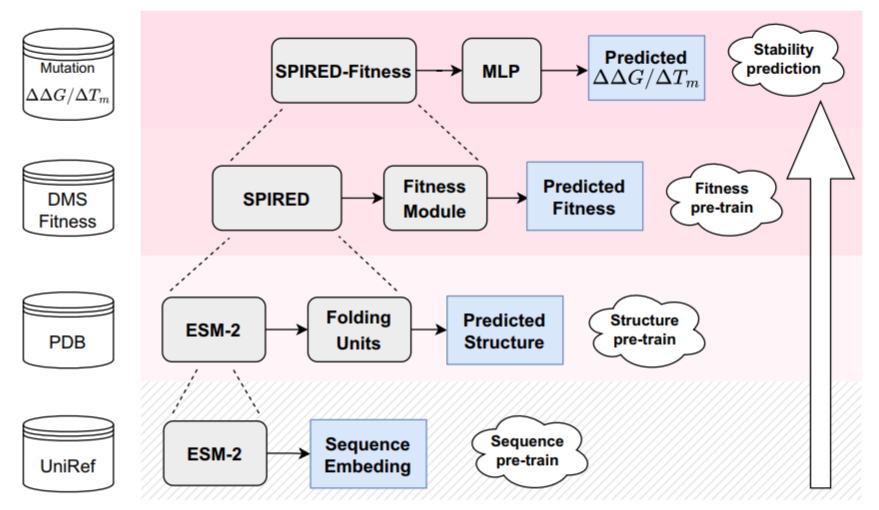
图6. 该研究中阶梯递进式的预训练策略
清华大学生命科学学院/北京生物结构前沿研究中心龚海鹏副教授为本文通讯作者;清华大学生命科学学院19级博士生陈应辉和19级博士生徐运昕为本文共同第一作者;清华大学生命科学学院本科生刘迪与邢耀光博士参与了本研究项目。本工作获得了国家自然科学基金委、国家科技部、北京生物结构前沿研究中心的经费支持。
参考资料
Yinghui Chen#, Yunxin Xu#, Di Liu, Yaoguang Xing and Haipen Gong*. SPIRED-Fitness: an end-to-end framework for the prediction of protein structure and fitness from single sequence. Nature Communications, 15(1):7400, 2024.
代码
https://github.com/Gonglab-THU/SPIRED-Fitness
SPIRED-Fitness网页服务
https://structpred.life.tsinghua.edu.cn/server_spired_fitness.html
SPIRED-Stab网页服务器
https://structpred.life.tsinghua.edu.cn/server_spired_stab.html
内容中包含的图片若涉及版权问题,请及时与我们联系删除

评论
沙发等你来抢