
DRUGAI
今天为大家介绍的是来自北京大学信息工程学院、化学生物学与生物技术学院省部共建肿瘤化学基因组学国家重点实验室、鹏城国家实验室合聘研究员和AI4S平台中心主任陈语谦教授团队与新加坡国立大学Lei Shen团队在Nature Communications上发表的论文,文章的第一作者为博士生杨梓铎和赵艺明。该团队开发了一种单步晶体结构优化方法,避免了在使用第一性原理计算进行晶体结构优化时的迭代过程,为大规模晶体结构优化和高通量计算提供了高效的解决方案。

背景介绍
原子/晶体结构优化在计算化学、计算物理和计算材料科学等领域具有广泛的应用。结构优化的主要目标是找到最低能量状态,也称为基态。基态结构是计算并预测其物理和化学性质的基础。该过程对表面化学反应、晶体缺陷调控以及药物设计等应用至关重要。
传统上,晶体结构优化常采用第一性原理计算方法,例如密度泛函理论(DFT)。在DFT计算中,每次迭代都涉及求解Kohn-Sham方程以确定电子密度分布,然后根据电子密度分布计算出系统的总能量和力场,系统中的原子在力场的引导下沿着降低系统能量的方向移动,如图1(a)所示。尽管基于DFT的结构优化方法精度较高,但其高计算需求和较差的可扩展性限制了其在大规模晶体结构优化和高通量计算场景中的实用性。
晶体结构数据库的快速发展使得通过数据驱动的方法来预测晶体基态结构成为了可能。机器学习(ML)方法为晶体结构优化提供了一条新路径,通过避免求解Kohn-Sham方程,显著降低了计算成本和时间。目前,采用ML方法进行晶体结构优化主要分为两类:迭代式ML方法和单步ML方法。迭代式方法通常使用图神经网络(GNN)作为代理模型,迭代地预测能量、力场和应力等物理量,如图1(a)所示,从而避免了求解计算量巨大的Kohn-Sham方程。然而,迭代式ML方法存在三个不足之处:第一,训练迭代式ML模型需要部分甚至全部DFT优化过程中的数据,且这些数据需要包含能量、力场和应力等物理量。目前大多数开源数据集并不包含优化过程的信息;第二,迭代式方法的迭代特点限制了其并行计算的能力;第三,此类方法无法完全避免迭代过程,因此仍然较依赖计算资源。相比之下,单步方法完全跳过了迭代步骤,直接从初始结构预测基态结构。然而,目前的单步方法仅适用于特定体系,通用性较差。
为了解决以上问题,论文提出了一种可拓展性强、通用性广的单步ML模型DeepRelax。DeepRelax绕过了传统计算中的迭代步骤,实现了单步计算预测晶体基态结构。进一步利用DFT对DeepRelax预测的结构进行优化,可实现快速收敛。在使用单个A6000 GPU进行计算时,DeepRelax仅需几百毫秒便能完成一个晶体结构的优化工作,与近期发表在《Nature Computational Science》上的M3GNet相比,速度提升了两个数量级。此外,DeepRelax还具备并行结构优化的能力,这一特点使其在高通量材料筛选和计算中的应用价值得到了显著提升。
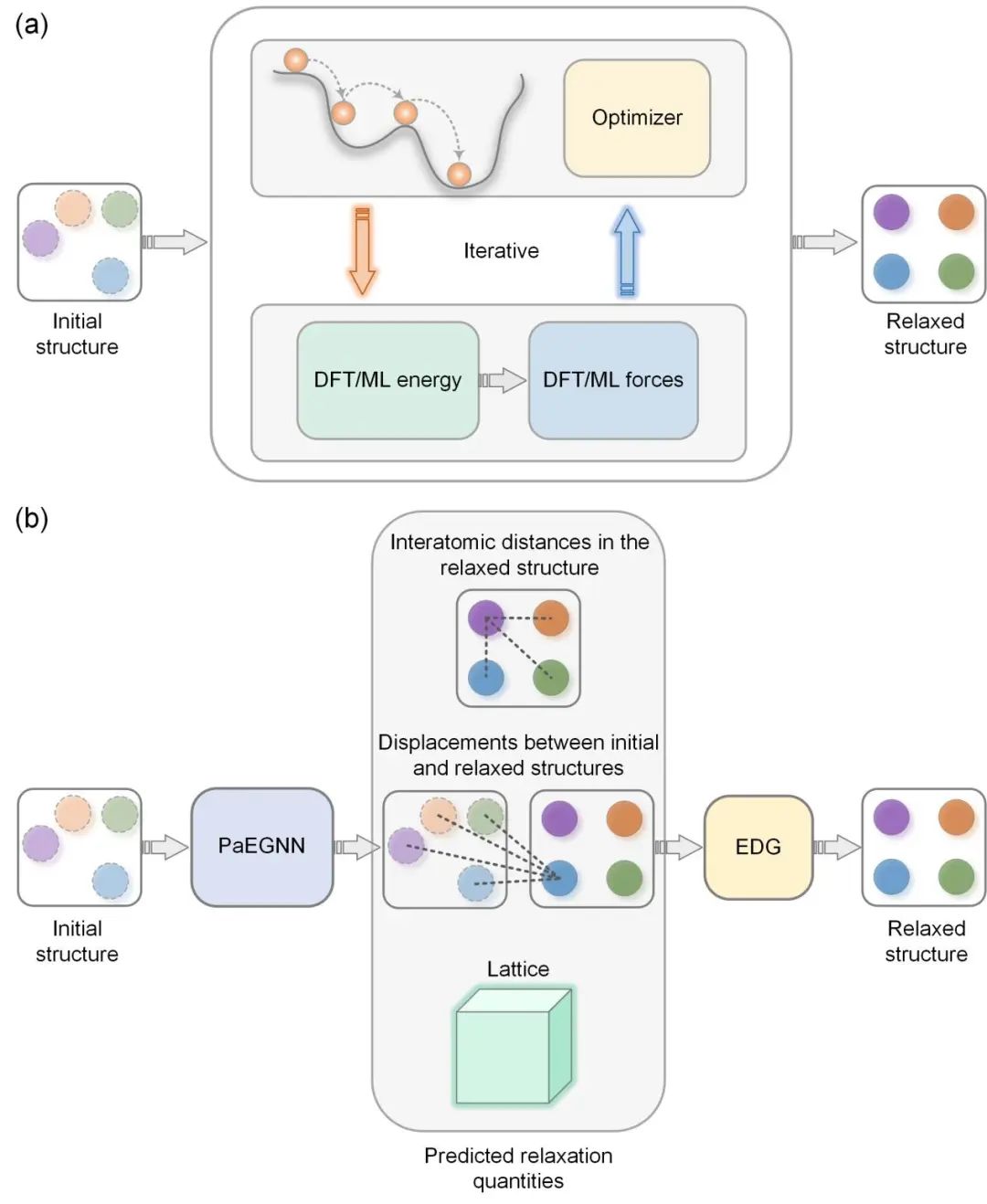
图1. 晶体结构优化方法(a)基于DFT和迭代式ML方法的晶体结构优化(b)论文提出的单步ML模型DeepRelax
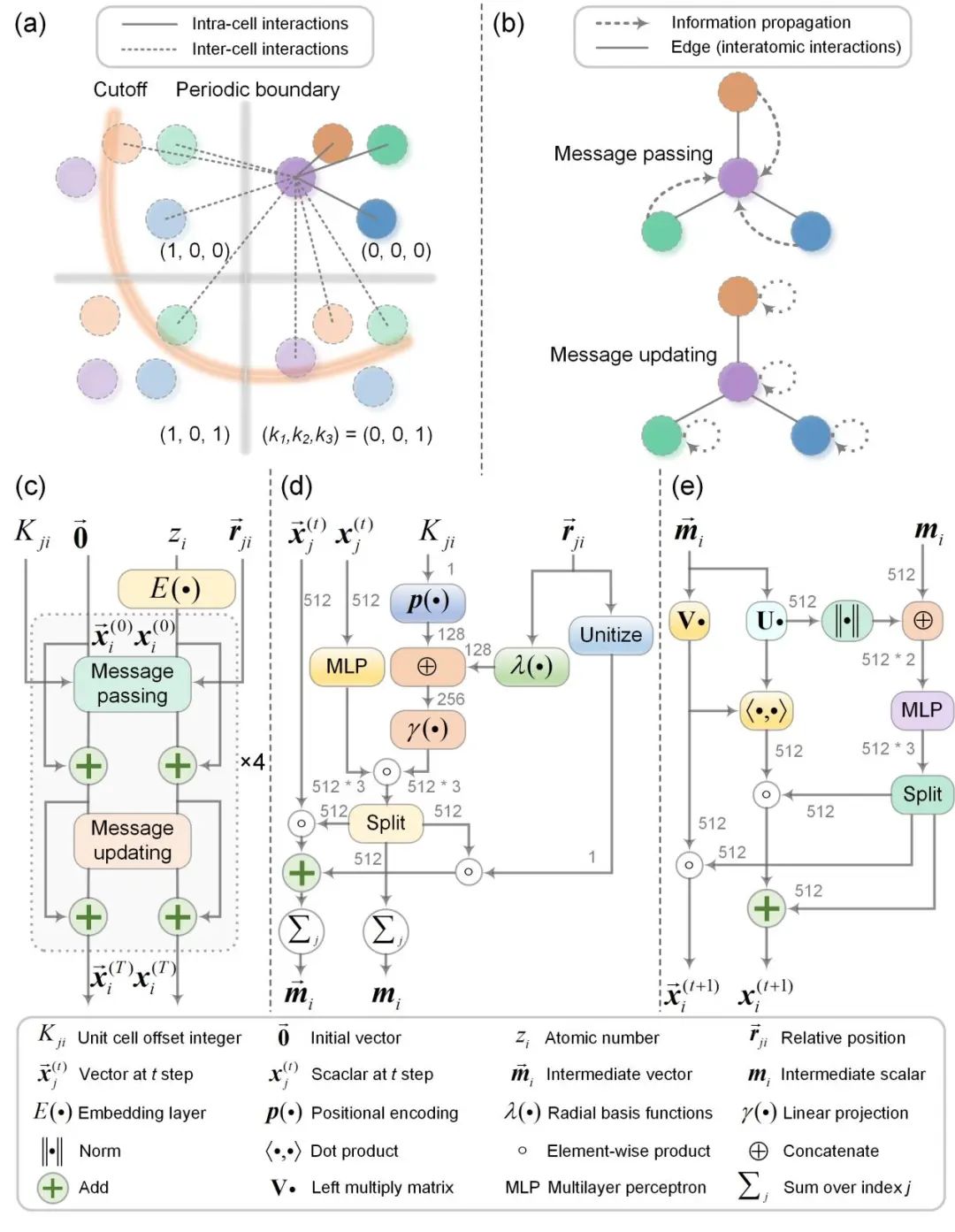
图 2. PaEGNN的核心原理(a)用于表示晶体结构的多边图(b)消息传递和消息更新(c)图卷积层的具体设计(d)消息传递的具体计算过程(e)消息更新的具体计算过程
模型架构
DeepRelax 的输入为初始或未优化的结构,随后通过一个周期性感知的等变图神经网络(PaEGNN)来预测基态结构的键长分布、优化前后的原子位移以及晶格常数,如图1(b)所示。通过这些预测的物理量,DeepRelax能够推导出基态结构的原子坐标。具体而言,论文提出了一种欧几里得距离几何求解器(EDG),该求解器基于预测的键长分布、原子位移和晶格常数,精确计算出满足这些物理量的原子坐标,从而获得基态结构,如图1(b)所示。
周期性感知的等变图神经网络(PaEGNN)
使用GNN进行晶体结构表征学习的第一步是将晶体结构转换成GNN可以处理的形式。晶体结构不同于常规的小分子化合物或生物大分子,它具有独特的周期性和对称性。因此,将晶体结构转换成图结构的时候(节点代表原子,边代表键),常常采用多边图表示法,即一个节点与另外一个节点可以形成多条不同的边,如图2(a)所示。
PaEGNN通过两个步骤逐步更新节点表示:消息传递、消息更新,如图 2(b) 所示。详细计算过程如图 (b)-(e) 所示。在消息传递阶段,每个节点通过接收其邻近节点的消息来模拟两体相互作用。消息更新阶段主要对每个节点内部的信息进行整合,以更新节点表示。
不同于常规GNN,论文构建的PaEGNN显式地考虑了晶体结构的周期性。具体而言,PaEGNN 对晶胞的偏移进行了唯一编码,从而对不同平移晶胞中的原子进行明确区分,以编码周期性边界条件。这一设计显著提高了结构预测的准确性。
不确定性估计方法
不确定性估计有助于评估模型预测结果的可靠性,对确保预测结果的可信度至关重要。若模型在预测结构的同时能够估计其预测结果的不确定性,则可以利用这些不确定性信息筛除不可靠的预测结果。DeepRelax同时估计内在不确定性(Epistemic Uncertainty)和外在不确定性(Aleatoric Uncertainty)。外在不确定性源于系统本身的随机性,而内在不确定性则由于模型对数据分布的理解不足所引起。
为估计外在不确定性,DeepRelax将预测的几何结构参数(包括键长和位移)视为拉普拉斯分布的位置信息,并额外预测相应的尺度参数。通过构建损失函数,使模型预测的几何结构参数符合拉普拉斯分布,从而使预测的尺度参数能够用于估计外在不确定性。对于内在不确定性,DeepRelax通过训练多个随机初始化的模型进行估计,利用模型间预测结果的差异来衡量不确定性。通过上述不确定性估计方法,DeepRelax能够更全面地评估模型预测结果的可靠性,从而在实际应用中显著提高模型的可信度。
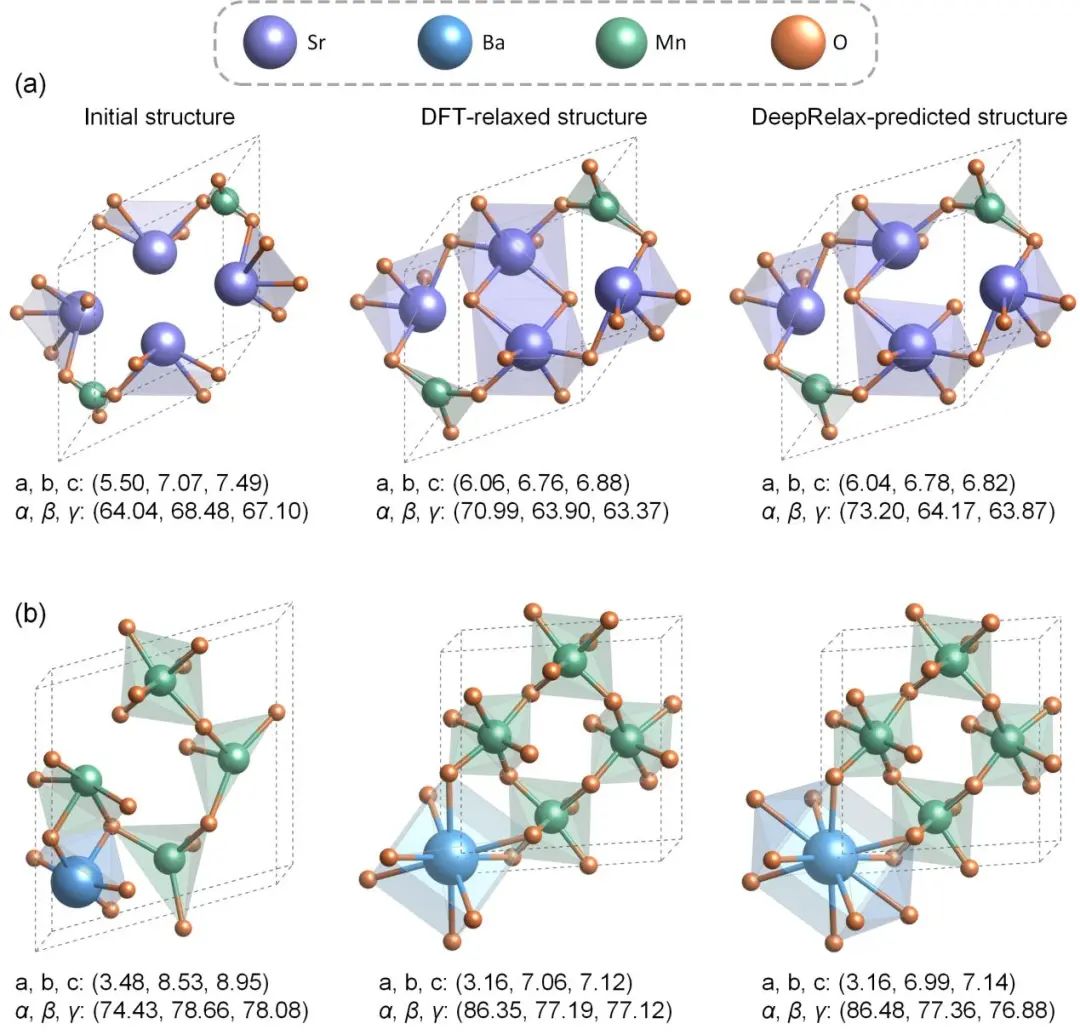
图 3. 使用DeepRelax进行结构优化的例子
结果
论文对DeepRelax进行了全面的验证,包括三维材料和二维材料在内的多个数据集,如X-Mn-O氧化物体系(X代表 Mg, Ca, Sr, and Ba)、Materials Project、C2DB、缺陷结构、范德瓦尔斯晶体。在X-Mn-O体系的测试中,DeepRelax预测的原子坐标以及晶格常数,相比未优化时分别提高了64.65%和71.49%,图3展示了两个使用DeepRelax进行结构优化的例子。即便面对元素类型丰富且结构多样化的Materials Project数据集,DeepRelax同样展现出了显著的性能,对比未优化结构,DeepRelax优化后原子坐标和晶格常数分别提高了超过30%和40%。对于二维材料数据集C2DB的测试也显示出相似的提升幅度。此外,论文还证明了DeepRelax适用于缺陷结构和范德瓦尔斯晶体。这些测试结果验证了DeepRelax的通用性,证明其适用于多种不同类型的材料体系。
DFT验证
为了进一步验证DeepRelax预测的基态结构在能量上的优势,论文以X-Mn-O氧化物体系、Materials Project和缺陷结构为例,对未优化结构(Unrelaxed)、DFT优化结构(Relaxed)以及DeepRelax预测的结构(Predicted)进行了DFT能量计算。如图4(b)和(d)所示,DeepRelax预测的结构能量显著低于未优化结构,并接近DFT优化结构的能量。这表明DeepRelax能够有效地找到低能量的结构。同时,将DeepRelax预测的结构作为初始结构进行DFT优化,可以显著减少达到基态结构所需的迭代步数,如图如图4(f)所示。这进一步证明了DeepRelax在加速DFT计算、提高计算效率方面的潜力。
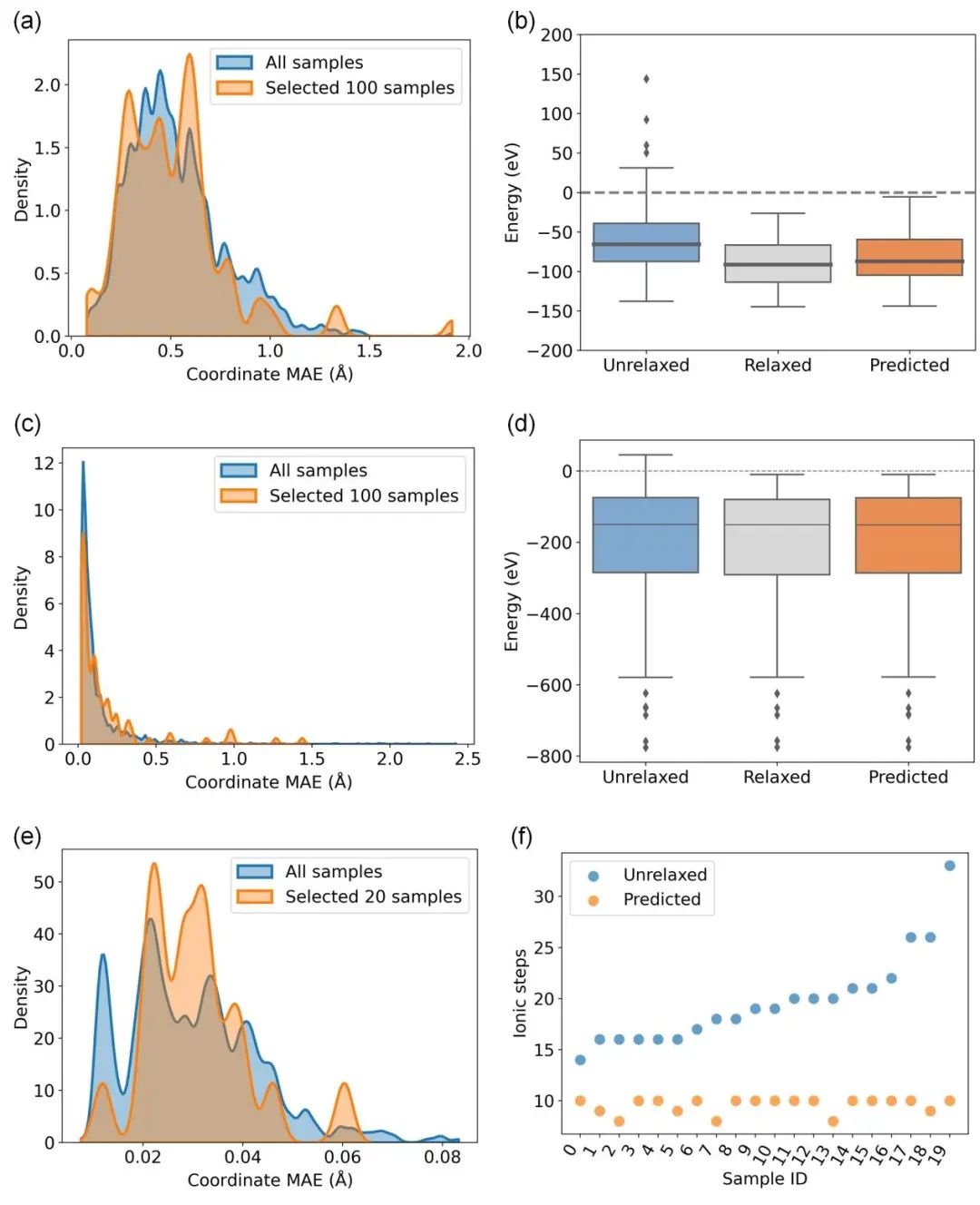
图 4. DFT验证结果
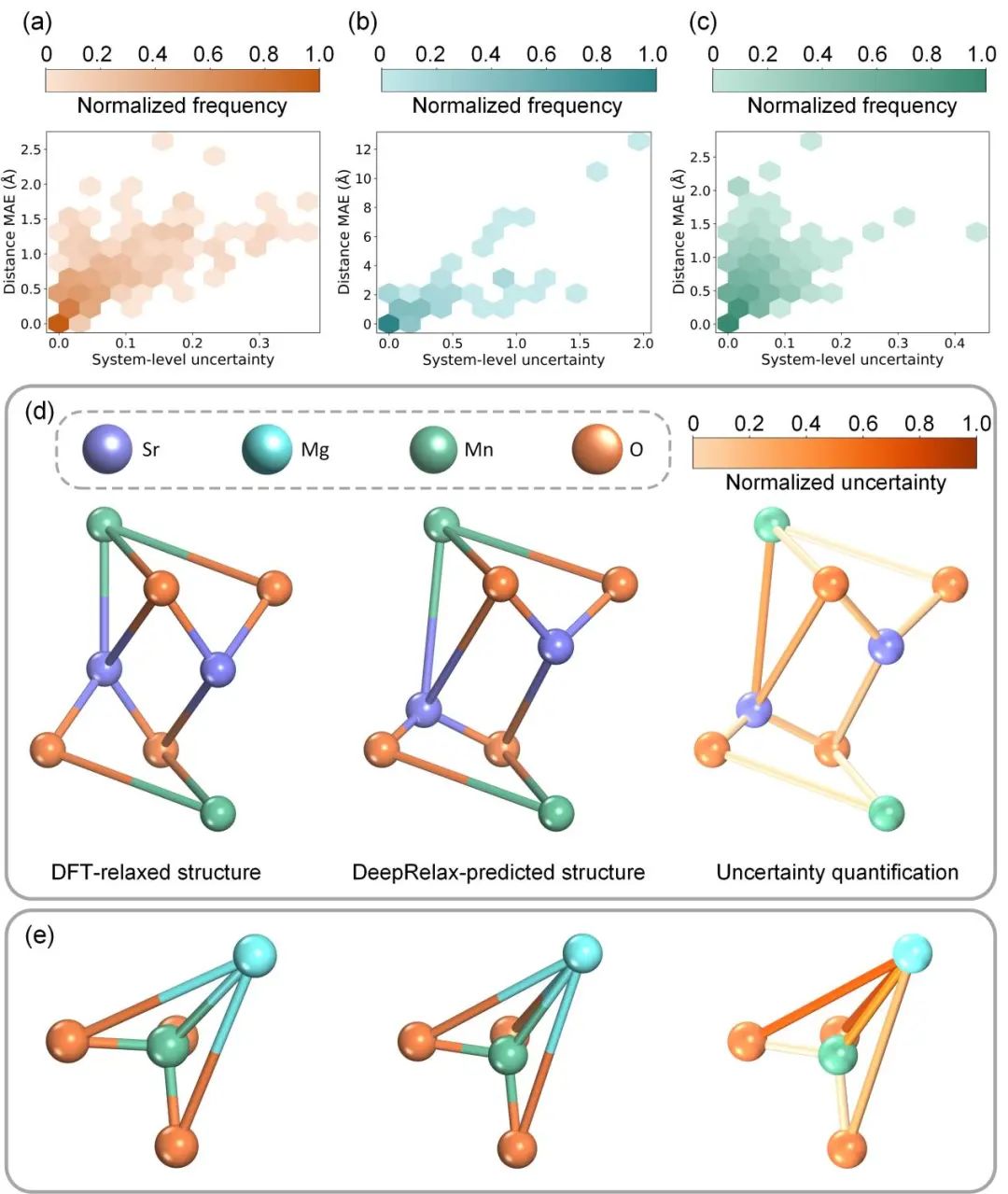
图 5. 不确定性估计
不确定性估计
为了验证论文提出的不确定性量化方法在反映模型预测置信度方面的有效性,作者计算了模型预测的键长误差与估计不确定性之间的斯皮尔曼秩相关系数。图5(a)-(c)分别展示了DeepRelax在X-Mn-O、Materials Project和C2DB数据集中,模型估计的不确定性与预测键长误差之间的关系。在这三个数据集中,估计不确定性与预测键长误差的相关系数分别为0.95、0.83和0.88,表明预测误差与估计不确定性之间存在高度正相关。这意味着不确定性估计能够有效反映模型预测的准确性。
此外,图5(d)-(e)展示了两个预测结构的不确定性可视化结果,进一步说明了预测键长误差与不确定性之间的相关性。这些可视化结果直观地显示了高不确定性区域通常伴随着较大的预测误差,反之亦然。
综上所述,这些结果表明,DeepRelax模型估计的不确定性能够作为评估预测结构准确性的可靠指标,从而提升模型在实际应用中的可信度和实用性。
总结
传统的晶体结构优化通常采用密度泛函理论(DFT)等第一性原理计算方法。尽管基于DFT的结构优化具有较高的精度,但其高计算需求和有限的可扩展性限制了其在大规模晶体结构优化和高通量计算中的应用。例如,近期Google DeepMind通过深度学习方法发现了220万个新的晶体结构(Nature, 2023, 624(7990): 80-85),若全部采用DFT进行优化,将消耗巨大的计算资源。针对这一挑战,DeepRelax提供了一种适用于大规模晶体结构优化的高效解决方案。
DeepRelax通过单步机器学习模型,能够快速预测接近基态的初始结构,显著减少了计算时间。随后,结合DFT进行精细优化,实现了高效与高精度的平衡。具体而言,DeepRelax模型在处理大规模数据时展现出优越的性能,不仅提高了计算效率,还保持了预测结果的精度。这一方法不仅适用于复杂的材料和化学系统,还支持并行GPU处理,进一步提升了计算速度。
通过将DeepRelax与传统的DFT方法相结合,可以有效解决高效性与高精度之间的矛盾,满足大规模晶体结构优化和高通量计算的需求。这不仅加速了材料发现和设计过程,还大幅降低了计算资源的消耗。总之,DeepRelax在晶体结构优化领域取得了显著进展,展示了其作为高效、可扩展且精准的工具,在材料科学研究中的巨大潜力。
参考资料
Yang, Z., Zhao, YM., Wang, X. et al. Scalable crystal structure relaxation using an iteration-free deep generative model with uncertainty quantification. Nat Commun 15, 8148 (2024). https://doi.org/10.1038/s41467-024-52378-3
代码
https://github.com/Shen-Group/DeepRelax
内容中包含的图片若涉及版权问题,请及时与我们联系删除

评论
沙发等你来抢