
DRUGAI
今天为大家介绍的是来自华盛顿大学Georg Seelig团队的一篇论文。蛋白质–蛋白质相互作用(PPIs)调控着许多细胞过程,且经过工程改造的PPIs在细胞和基因治疗中具有应用价值。作者在此介绍了一种大规模并行的PPI测量方法——MP3-seq,这是一种简单易用且高度可扩展的酵母双杂交方法,用于测量PPIs。在MP3-seq中,DNA条形码(barcode)与特定的蛋白质对关联,条形码的富集情况可通过测序读取,从而直接测量相互作用的强度。作者展示了MP3-seq的高度定量特性,并可扩展到超过10万个相互作用。作者应用MP3-seq对经过合理设计的异源二聚体家族间的相互作用进行了表征,并研究了什么元素能够给coiled-coil相互作用添加特异性。最后,作者使用AlphaFold-Multimer(AF-M)预测螺旋异源二聚体的结构,并在基于物理的能量项上训练线性模型以预测MP3-seq的数值。研究发现,基于AF-M的模型对预筛选相互作用有一定价值,但定量排名其强度仍需要实验测量。

合成蛋白结合体能够介导蛋白质或细胞间的相互作用,有望在细胞治疗、合成生物学和材料科学等领域引发革命性的变化。早期研究通常依赖于天然的相互作用结构域,如SH3和PDZ。然而,这些组件由于存在交叉干扰,不适合用于理性设计大规模的组装。因此,合成蛋白电路依然比生物蛋白–蛋白相互作用(PPI)网络要小得多且更简单。为了扩大合成蛋白电路的规模,需要大规模的模块化相互作用结构域库。理想情况下,这些相互作用结构域应是正交的,即每个结构域只与其指定的结合伙伴相互作用。
通过理性设计异源二聚体来满足这一需求已取得了令人鼓舞的结果,但要创建大型且完全正交的二聚体集合仍然充满挑战。
许多PPI测量方法已经被开发出来,可以用于运行不同通量水平的全互作PPI筛选。酵母双杂交(Y2H)方法是表面展示技术的强大替代方案,用于表征蛋白质–蛋白质相互作用(PPIs)。在Y2H中,一个蛋白与DNA结合域(DBD)融合,另一个蛋白与转录激活域(AD)融合。如果两者发生相互作用,就会重新组装成一个功能性的转录因子,驱动生长必需酶的表达。早期的Y2H方法通过基于平板的筛选测试少量PPI,但实验室自动化和混合策略使得蛋白质组规模的筛选成为可能。为进一步解决Y2H的扩展问题,开发了高通量Y2H(HT-Y2H)和酶互补方法,这些方法利用新一代测序读取相互作用强度。同时,为分析HT-Y2H数据,开发了定制的工作流程。大多数实验方法需要在大肠杆菌中构建文库或依赖于酵母交配,因此需要分别将蛋白质文库转化到MATα和MATa型酵母中。已经开发了高通量的细菌双杂交测定法,为PPI筛选提供了一种非真核生物的替代方案,避免了酵母交配和文库转移。然而,在测试蛋白质相互作用时,类似于其天然环境的条件是理想的。因此,使用基于Y2H的测定法测试天然来源于真核生物的胞内蛋白的相互作用是有益的。特别是,在酵母中表达这些蛋白相较于在细菌中表达,可以获得更准确的折叠、引入翻译后修饰以及提高溶解度。然而,在测试蛋白质相互作用时,类似于其天然环境的条件是理想的。
深度学习模型如AlphaFold2(AF2)和RoseTTAFold已经显示出能够以接近实验精度预测蛋白质结构。对于多链蛋白的结构预测,可以使用原版AF2模型或专门的模型如AF-Multimer(AF-M)。考虑到这些模型的运行速度远快于酵母双杂交(Y2H)测定法,探索如何利用这些模型对PPI进行预筛选或补充PPI测定非常具有吸引力。已有工具开发用于以全互作方式运行AF-M,以辅助PPI预筛选,并且AF-M的误差指标已被证明是最先进的蛋白–肽相互作用预测器。然而,AF-M在预测蛋白结合正交性方面的能力仍不明确。
MP3-seq工作流程
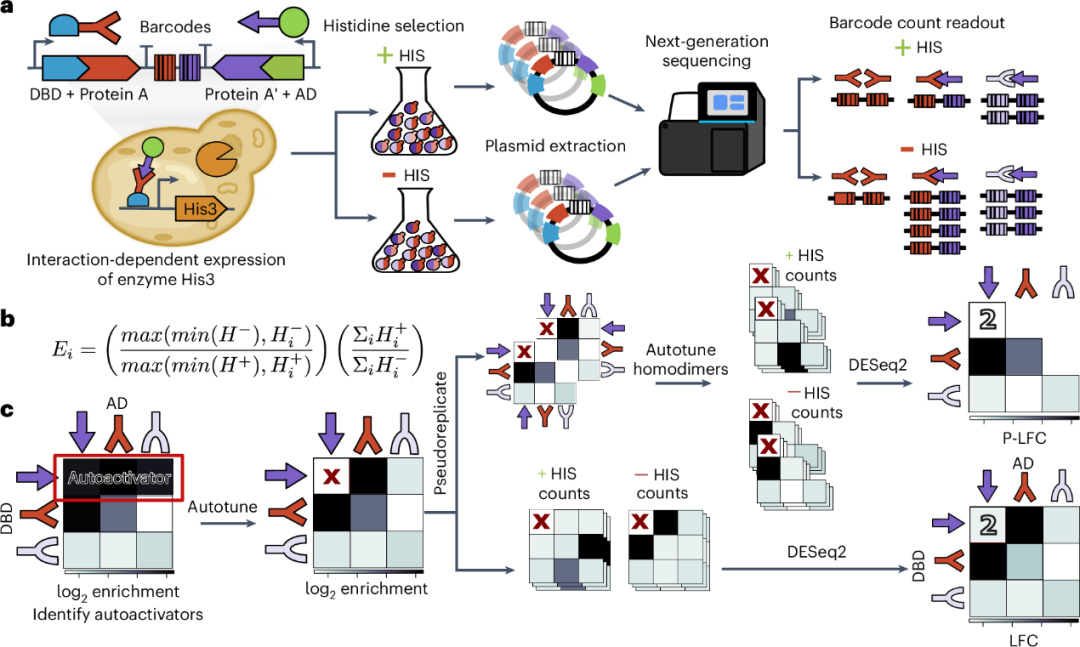
图 1
在MP3-seq中,测量两种特定蛋白相互作用所需的所有分子组件都编码在一个质粒上(图1a)。质粒文库通过在酵母中直接进行同源重组构建,以测量一组蛋白质的所有可能相互作用。首先,将DNA片段混合物转化到单倍体MATa型酵母中。一个片段是含有着丝粒(CEN)序列、选择标记、DBD和AD的主链。作者使用小鼠转录因子Zif268的Cys(2) His(2)锌指域作为DBD及其对应的启动子来驱动生长必需酶His3的表达。源自单纯疱疹病毒的VP16蛋白域被用作AD。其他片段包含一个目标蛋白及其相关条形码,两者由终止子序列分隔。由于其短小且独特的序列,作者在大多数实验中使用了Tsynth23和Tsynth27作为终止子。
转化后,作者在不含色氨酸的培养基中进行延长生长和筛选步骤,以确保质粒的维持。在此阶段,CEN序列确保每个酵母细胞(以及后续的子细胞)大约含有一个文库质粒,这对于将转染酵母的生长与条形码计数关联至关重要。部分细胞被冷冻保存,其余的细胞在缺乏组氨酸(His)的培养基中进行筛选。从组氨酸预筛选和后筛选的细胞中提取质粒DNA,并对含条形码的区域进行扩增(图1a,右侧)。条形码扩增产物被测序,通过条形码计数计算其相对富集情况,作为相互作用强度的指标(图1b)。由于一些蛋白与AD或DBD融合可能会影响其表达或折叠,因此作者对所有蛋白进行了两种融合顺序的测试(DBD与P1融合、AD与P2融合;DBD与P2融合、AD与P1融合)。
MP3-seq分析流程
首先,作者计算了PPI融合顺序在组氨酸预筛选和后筛选阶段之间的富集情况(图1b)。接着,检测自激活蛋白(autoactivators),并将其组氨酸后筛选的值替换为从非自激活融合顺序(nonautoactivating fusion order)计算的值(详见方法中的Autotune);自激活是Y2H实验中的一种错误模式,表现为某个蛋白质对所有相互作用伙伴都显示出高富集,暗示选择标记的表达被非特异性激活。通常,这种行为只出现在AD或DBD其中一种融合方向,而不会同时出现在两种方向中。如果发现自激活蛋白,其同源二聚体将无法通过Autotune纠正,因为没有可用于校正的反向融合顺序。
去除自激活蛋白后,可以将重复实验的富集值平均,得到相互作用强度值。或者,为了校正由于不同测序深度、选择时间和其他实验因素导致的“实验重复间读数分布”(read count distribution between experimental replicates)的差异,作者使用DESeq2软件包计算log2倍数变化(LFC)。DESeq2计算多个重复间的差异富集,并提供经过Hochberg调整的Wald检验P值(Padj),用于识别LFC显著不同于零的PPI。
在某些情况下,将MP3-seq测量的两种融合顺序的结果结合起来是理想的(例如,当将MP3-seq的LFC与目标对的Kd值进行比较时)。作者将每种融合顺序视为独立的一组测量,将它们转换为两个伪重复。然后,将这些伪重复与DESeq2结合计算LFC(图1b)。作者将这些值称为伪重复LFC(P-LFC)。
MP3-seq 基准测试使用正交的卷曲线圈二聚体
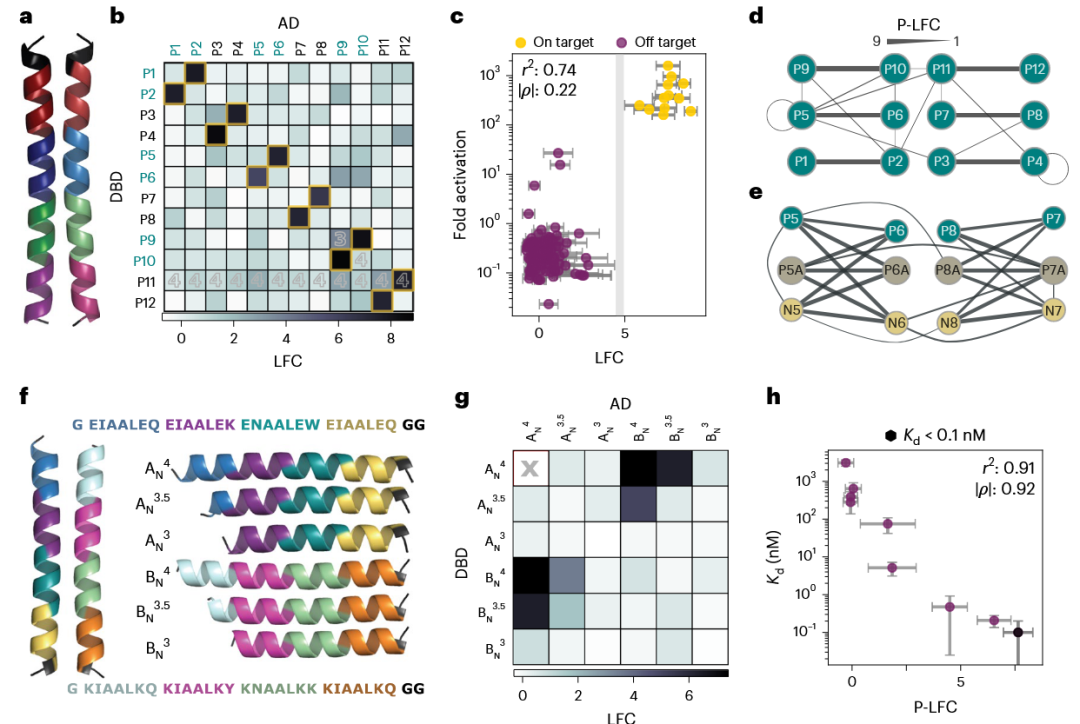
图 2
为了验证 MP3-seq,作者筛选了NICP数据库中的六个正交 1×1 的 144 对相互作用(图2a)。用了包含12种蛋白质的24个DNA片段,每个蛋白质分别与AD或DBD融合。所有相互作用对在混合实验中组装,并使用MP3-seq量化了相互作用强度。如预期的那样,组氨酸预选择条形码计数随着时间保持稳定,并且应用不同水平的3-氨基-1,2,4-三唑(3-AT,His3的竞争性抑制剂)会影响组氨酸后选择计数。从五次实验重复中计算了两种取向的LFC,几乎所有相互作用都发生在设计的(靶向)合作伙伴之间(即P1:P2, P3:P4等;图2b)。在MP3-seq中,同源二聚体质粒的组装效率较低,通常导致输入条形码计数较低,但覆盖范围足以纳入分析。这种现象可能是由于增加的序列同源性干扰了质粒的组装。为了进行更定量的验证,作者将MP3-seq的LFC与HEK293T细胞中萤火虫荧光素酶表达实验的相互作用得分进行了相关分析,发现一致性较好(r2 = 0.74;图2c)。图2d将这些PPI的P-LFC MP3-seq数据表示为图,其中每个蛋白质为一个顶点,边缘是由P-LFC加权的显著相互作用。
为了评估MP3-seq的数值是否能定量反映相互作用强度,作者对设计用于覆盖广泛Kd值范围的不同长度1×1的相互作用进行了三次全交互重复实验(图2f)。这些相互作用预计并非正交,因为通过用半个或整个七肽来截断两个母体四重七肽(parent four-heptad)结合体(AN 4, BN 4)来实现较弱的相互作用(图2g)。MP3-seq的P-LFC与先前测量的解离常数在大约三个数量级范围内高度相关(r2 = 0.94;图2h)。
编译 | 黄海涛
审稿 | 王梓旭
参考资料
Baryshev, A., La Fleur, A., Groves, B., Michel, C., Baker, D., Ljubetič, A., & Seelig, G. (2024). Massively parallel measurement of protein–protein interactions by sequencing using MP3-seq. Nature Chemical Biology, 1-10.
内容中包含的图片若涉及版权问题,请及时与我们联系删除

评论
沙发等你来抢