
DRUGAI
今天为大家介绍的是来自瑞士的Vassily Hatzimanikatis与Ljubisa Miskovic团队的一篇论文。生成大型组学数据集已经成为获取细胞过程洞察的常规手段,但解读这些数据集以确定代谢状态仍然具有挑战性。动力学模型可以通过明确关联代谢物浓度、代谢通量和酶水平来帮助整合组学数据。然而,确定细胞生理背后的动力学参数为这些代谢数学模型的广泛应用带来了显著的障碍。在此,作者介绍了RENAISSANCE,这是一种生成式机器学习框架,可以高效地为具有符合实验观察的动态特性的大型动力学模型进行参数化。通过无缝整合各种组学数据和其他相关信息,包括细胞外培养基成分、物理化学数据和领域专家的专业知识,RENAISSANCE能够准确表征大肠杆菌的细胞内代谢状态。它还能估计缺失的动力学参数,并将其与稀疏的实验数据相协调,从而显著减少参数的不确定性并提高准确性。该框架对于研究涉及代谢物和酶水平变化以及酶活性变化的代谢变异的健康和生物技术领域的研究人员将非常有价值。

生物技术和健康科学的进步高度依赖于整合高通量技术产生的数据,以获得对细胞过程的深入理解。基因组规模模型作为代谢信息的数学表示,通过考虑遗传和物理化学约束来整合组学数据。然而,这种方法在确定细胞内代谢状态时存在很大不确定性,尤其是在确定精确的代谢物水平和反应速率方面。
代谢动力学模型可以通过整合代谢组学、通量组学、转录组学和蛋白质组学数据来解决这一问题。与基于约束的模型不同,动力学模型还能够捕捉代谢的时间依赖性反应。这使得动力学模型在解决复杂生物医学现象和生物技术问题方面具有很大潜力,如肿瘤微环境中的代谢重编程、癌症与代谢和生物钟的关系、药物吸收与代谢动态等。
尽管动力学模型具有整合数据和识别与表型相关的代谢特征的能力,但其应用仍然受到限制,主要挑战在于缺乏细胞内动力学参数的精确知识。构建这些模型往往需要复杂的计算和专家的深入参与,因此难以用于多种生理条件下的研究。
RENAISSANCE是一个机器学习框架,在无需训练数据的情况下能够高效地为代谢动力学模型参数化。参数化动力学模型的行为是高度非线性的,为了捕捉这种非线性行为,作者使用了复杂度相当的前馈神经网络,并使用自然进化策略对其进行优化,以获得具有期望特性的动力学模型。该模型能够大大减少传统动力学建模所需的计算时间。最后,作者通过包括生成大肠杆菌代谢的动态模型群、准确表征其细胞内代谢状态以及整合实验数据这三项研究展示了其应用。
为什么要预测细胞内代谢状态?
预测细胞内代谢状态对于理解细胞的功能和行为、关联代谢特征与疾病表型以及开发新的治疗策略至关重要。它可以帮助识别疾病的代谢特征,优化代谢工程,提高药物设计和个性化医疗的效率。
使用生成式模型有什么好处?
参数估计:生成式模型可以估计缺失的动力学参数,并与稀疏的实验数据相结合,从而减少不确定性,提高模型的准确性。
效率:相比传统的动力学建模方法,生成式模型大大加快了模型构建的过程,减少计算时间。
广泛适用性:生成式模型可以整合多种组学数据和外部信息,生成符合生物学实验观察的模型,可用于不同的生理条件和生物化学属性研究。
为生物学相关的动力学模型参数化
作者开发了RENAISSANCE,这是一个用于生物学相关动力学模型参数化的机器学习框架。生成的模型与实验观察到的稳态相一致,并产生与实验观察相匹配的动态代谢反应时间尺度。
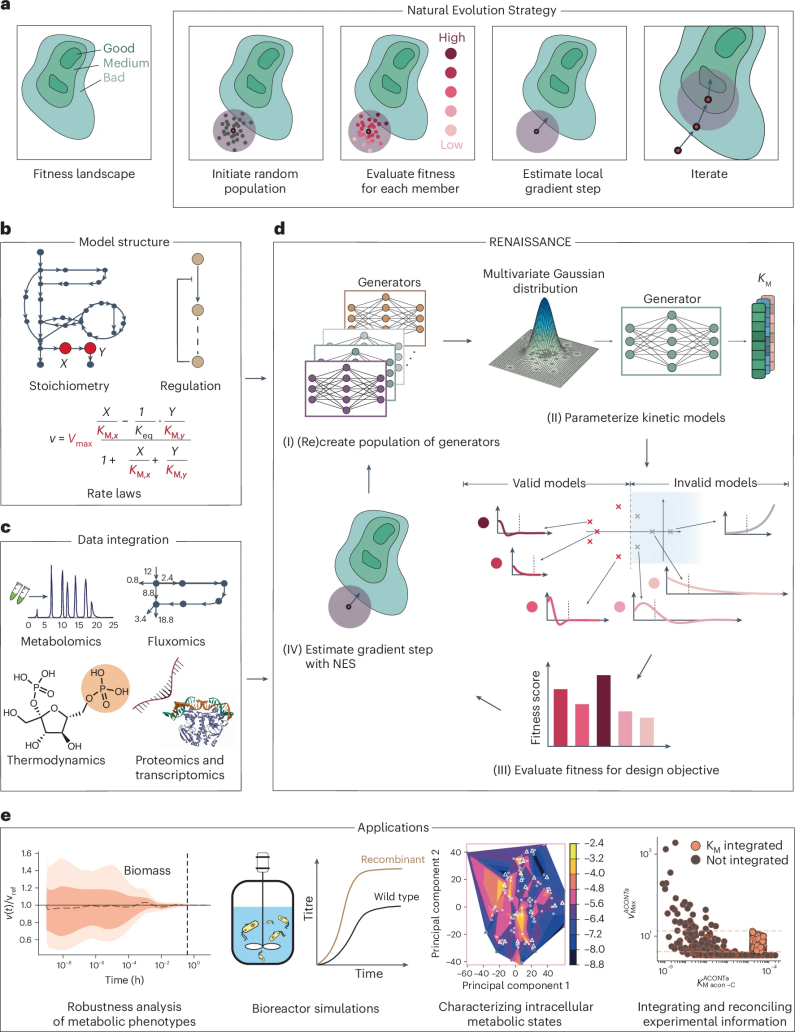
图 1
如图1b, c所示,RENAISSANCE的输入是代谢物浓度和代谢通量的稳态分布,它通过整合代谢网络的结构属性(化学计量、调控结构和反应速率定律)和可用数据(包括代谢组学、通量组学、热力学、蛋白质组学和转录组学)来计算。
RENAISSANCE使用前馈神经网络(生成器)来对动力学模型进行参数化,生成器网络的大小由动力学模型的复杂性决定。如图1d所示,利用自然进化策略(NES),它通过四个迭代步骤优化生成器的权重,直到生成生物学相关的模型:
步骤Ⅰ:随机初始化一组生成器的权重。使用多个生成器有助于更全面、更高效地探索参数空间。每个生成器接受多变量高斯噪声作为输入,并生成一组与网络结构和整合数据一致的动力学参数。
步骤Ⅱ:将这些参数集用于参数化动力学模型。
步骤Ⅲ:通过计算模型的雅可比矩阵的特征值及其对应的主时间常数来评估每个参数化模型的动态性。基于此评估,对生成器进行奖励分配。
步骤Ⅳ:每个生成器的奖励被反馈给NES,以找到性能最好的生成器。随后,NES通过向最优生成器的权重注入预定义的噪声水平对其进行突变,从而重新创建一组生成器(步骤Ⅰ)。
重复步骤Ⅰ–Ⅳ,直到获得满足用户定义设计目标的生成器,例如最大化生物学相关动力学模型的出现。如图1e所示,该生成的动力学模型具有多样性,可应用于广泛的代谢研究。
生成大肠杆菌代谢的大规模动力学模型
作者研究了产生蒽吲哚酸的大肠杆菌菌株W3110 trpD9923,以测试和验证RENAISSANCE。该菌株的动力学模型结构采用自Narayanan等人的模型,包含113个非线性常微分方程,由502个动力学参数参数化,包括384个米氏常数(KM)。它涵盖了123个反应,并描述了核心代谢途径,包括糖酵解、戊糖磷酸途径(PPP)、三羧酸循环(TCA)、补偿反应、莽草酸途径、谷氨酰胺合成和生长的汇总反应。目标是找到与实验观察到的该大肠杆菌菌株的134分钟倍增时间一致的动力学参数。
作者使用基于热力学的通量平衡分析来整合实验数据,并计算5000个代谢物浓度和通量的稳态剖面。作者选择其中一个剖面作为RENAISSANCE的输入,并确定了一组超参数,使三层生成器神经网络的框架性能最佳。
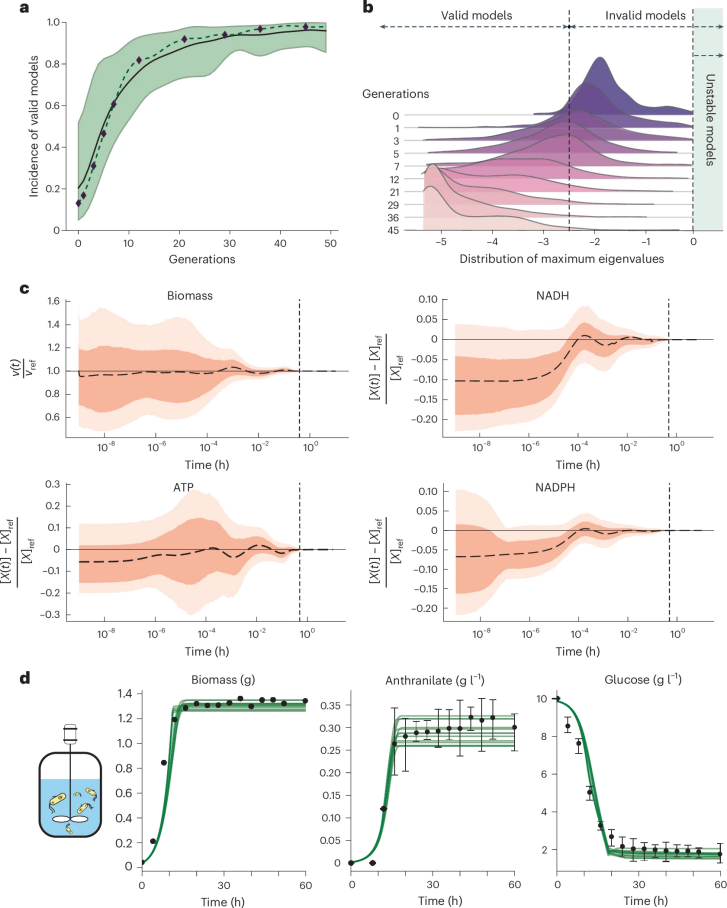
图 2
如图2a所示,为了对生成的模型进行进一步分析,作者选择了一个快速收敛的统计重复(虚线),并从该重复中选择了10个发生率随代数单调递增的生成器(黑色菱形)。对于每个选定的生成器,作者生成了500组动力学参数并检查了所得最大特征值的分布(图2b)。生成的模型在优化过程中逐渐从具有慢动力学(λmax > -2.5)转变为具有快动力学,代谢过程在随后的细胞分裂前稳定下来,这表明RENAISSANCE生成的模型能够捕捉实验观察到的动力学。
由于细胞生物在受到扰动时保持表型稳定性,作者生成的描述细胞代谢的模型也应具备这种特性。为了测试模型的稳健性,作者将稳态代谢物浓度扰动至±50%,并验证扰动系统是否回到稳态。如图2c所示,对归一化生物量随时间变化的检查表明,在24分钟内,100%的扰动模型的生物量恢复到参考稳态(v(t)/vref = 1)。整体检查所有胞质代谢物显示,75.4%的模型在24分钟内恢复到稳态,93.1%在34分钟内恢复,表明生成的动力学模型具有稳健性,并遵循所施加的特定生物物理时间尺度约束。
接下来,作者在非线性动态生物反应器模拟中测试了生成的模型,这些模拟紧密模仿了真实实验条件。图2d中生物量生产的时间演化显示出与典型实验观察相似的趋势,包括大肠杆菌生长的明显指数和稳定期。同样,葡萄糖摄取和蒽吲哚酸生产也再现了实验中观察到的趋势,其中葡萄糖消耗停止,蒽吲哚酸生产在约20小时达到饱和。这项研究表明,RENAISSANCE模型即使没有隐式训练以再现发酵实验,也能准确再现细胞代谢的生理学可观察和新兴特性。
表征大肠杆菌的细胞内代谢状态
准确确定代谢物浓度和代谢反应速率对于将代谢特征与表型关联至关重要。然而,我们在确定细胞内代谢状态方面的能力有限。即使有不断增加的生理学和组学数据,细胞内状态仍存在相当大的不确定性。作者建议使用动力学模型来减少这种不确定性,因为它们能够明确耦合酶水平、代谢物浓度和代谢通量。此外,动力学模型还允许我们在稳态数据之外考虑动态约束,从而进一步减少不确定性。
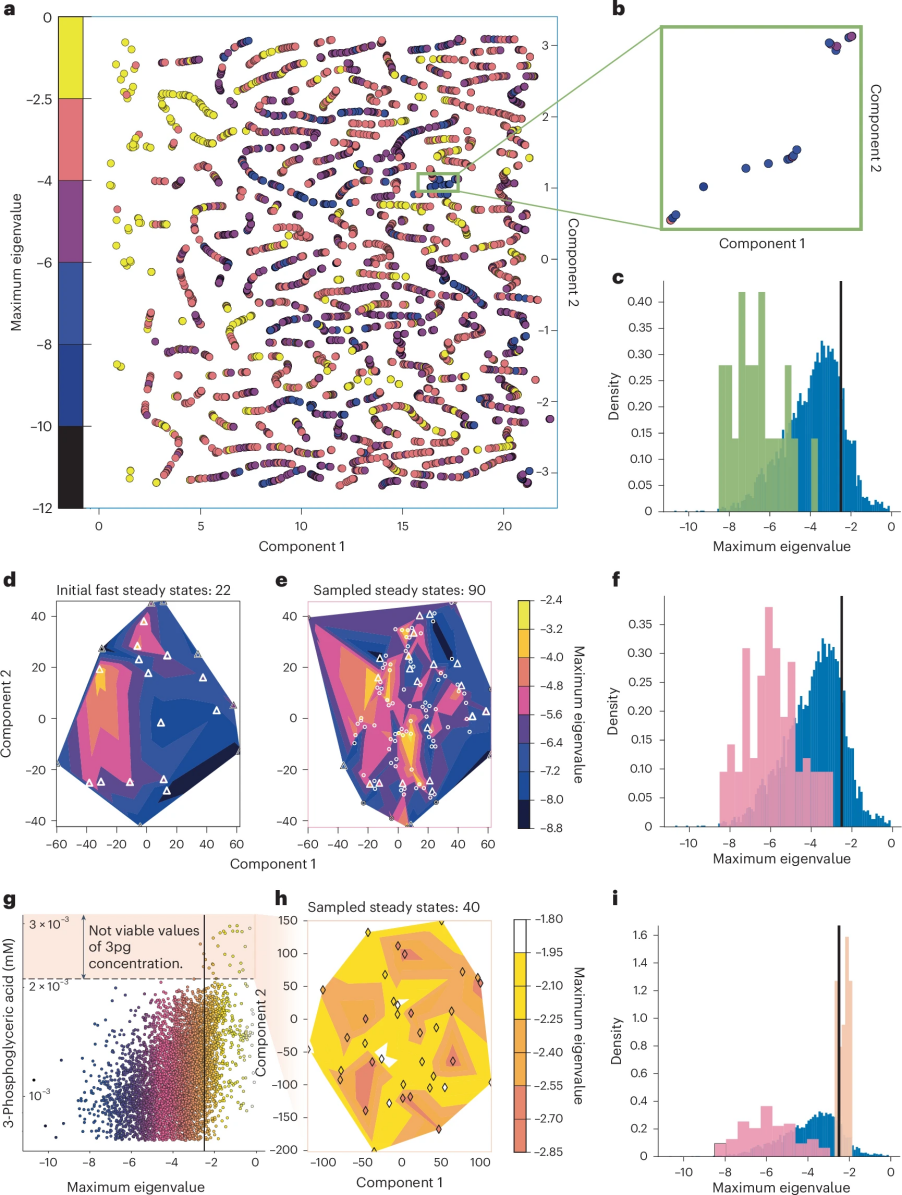
图 3
作者从这个不确定空间中采样了5000个代谢物浓度和代谢通量的稳态剖面,并使用RENAISSANCE为每个稳态找到最快的可能动态(最大负特征值,λmax)。作者通过主成分分析(PCA)和t分布随机邻嵌入(t-SNE)对稳态剖面进行降维并可视化,根据得到的λmax对每个稳态剖面进行着色(图3a)。作者观察到研究的稳态剖面的动力学存在很大的变化。在5000个稳态剖面中,有918个(18.4%)的λmax大于-2.5,这意味着这些细胞内代谢状态不符合实验观察。
由于t-SNE在将点从高维空间投影到低维空间时优化了点之间的局部距离保留,作者假设从包含与快速动力学相关的稳态剖面(蓝色点)区域中采样将产生满足动态要求的稳态剖面。相反,从与慢动力学对应的相邻剖面区域(黄色点)采样可能导致剖面不满足动态要求。
为了测试这个假设,作者选择了这些稳态剖面区域中的一个,它包含22个具有快速动力学的稳态(-3.8 ≤ λmax ≤ -8.5)。作者从该邻域的高斯分布中采样了90个附加稳态,其中均值和标准差基于初始的22个稳态估计。图3d-f中采样的稳态的线性化动力学与初始的22个稳态处于相同范围内,这证实了作者的假设。因此,RENAISSANCE能够选择与实验观察到的动力学一致的细胞内状态子集,并生成具有相同特征的其他状态。
接下来,作者检查了5000个稳态剖面的各个代谢物浓度,以识别与实验观察到的表型相对应的模式。作者观察到某些代谢物的浓度与动力学之间存在明显的偏差(图3g)。例如,对于3-磷酸甘油酸,仅在该代谢物浓度低于约0.002 mM时获得具有相关动力学的模型。相比之下,3-磷酸甘油酸浓度在0.002至0.003 mM之间的稳态剖面不具备相关动力学。图3h, i证明,几乎所有在支持浓度之外的新细胞内状态都未产生相关动力学的模型。这一结果表明,源自动态响应的信息可用于将细胞内代谢物的值约束在特定范围内。
总体而言,对广泛的细胞内状态进行动态表征可以减少稳态剖面、单个代谢物浓度和代谢通量方面的不确定性。
整合和协调实验信息
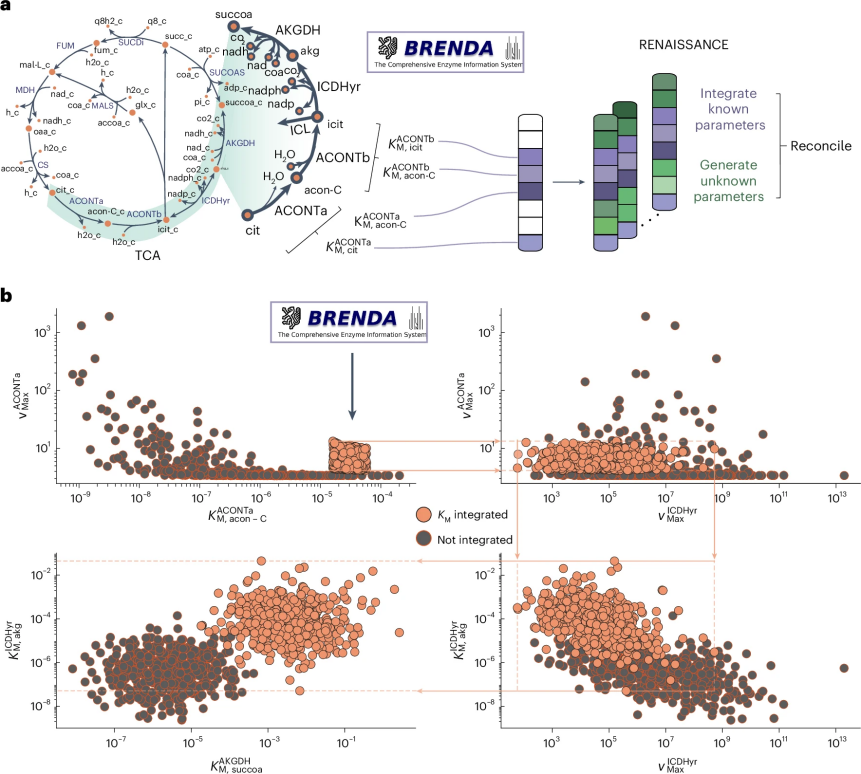
图 4
为了研究整合的动力学数据如何约束未知的动力学参数,作者首先整合了TCA循环中柠檬酸酶(ACONTa,b)的四个KM值(图4a),得到了具有高有效模型发生率(>99%)的生成器,并生成了500个有效的动力学模型。为了量化整合一个实验KM值对其他动力学参数的生成值的影响,作者将其他KM和最大速度(vmax)的估计值与没有整合任何动力学参数时得到的估计值进行了比较。
如图4b所示,在反应水平上整合柠檬酸酶的KM值限制了vACONTamax的估计值。由于整个网络中vmax值的相关性,通过KM整合限制vACONTamax的估计范围也限制了其他最大速度的估计范围,如vICDHyrmax。这种限制进一步影响了网络中的下游KM值,如KICDHyrM,akg和KAKGDHM,succoa。这些结果表明,仅整合局部于一个酶(ACONTa,b)的小量实验数据,会在整个代谢网络中传播并改变其他动力学参数。
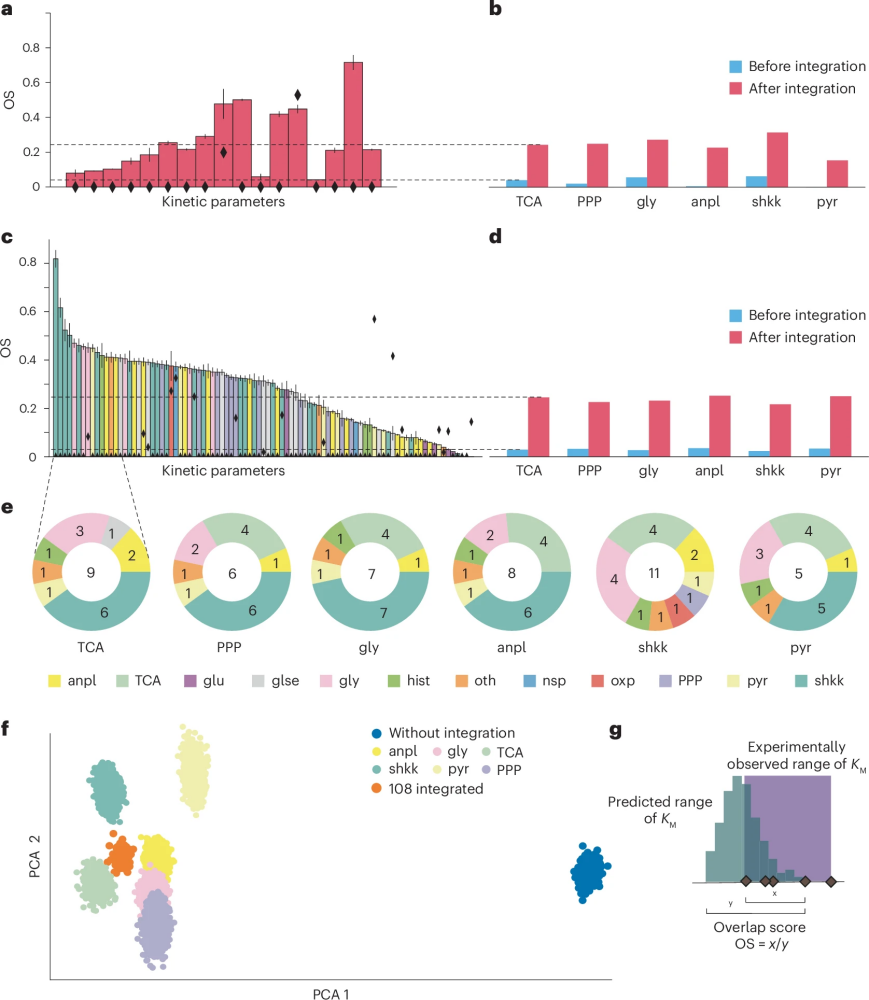
图 5
接下来,作者探讨了随着整合的实验KM值数量的增加,RENAISSANCE是否能改进KM的估计值。通过实验观察和RENAISSANCE估计的TCA KM值范围之间的重叠得分(OS)进行量化比较显示,与未整合任何KM值的情况相比,整合KM值改善了同一子系统内未整合的各个KM值的估计(图5a, 红条)。与未整合实验KM值的情况相比(图5b, 蓝条)整个子系统的平均预测准确性也有所提高(红条)。对于其他子系统,如PPP、糖酵解、补偿反应、莽草酸途径和丙酮酸代谢,作者进行了类似的分析,结果一致:整合实验信息后,同一子系统内KM值的估计都得到了改善(图5c, d)。这些发现表明,整合实验信息可以提高预测准确性,超越子系统级别。
作者进一步研究了整合实验动力学数据对缺乏可验证实验测量的参数的影响,这些参数占384个KM中的276个。为了对整合的影响进行定性评估,作者使用PCA可视化了RENAISSANCE对这些未知KM的预测(图5f)。分析显示,当整合实验数据时,这些KM的估计值发生了显著变化,而没有整合数据时则没有(蓝色簇)。此外,整合实验数据的情况下的估计比没有整合的更相似。
这些结果表明,整合实验动力学信息可以减少细胞内代谢状态的定量不确定性,使RENAISSANCE能够对整个代谢网络的动态特性做出更有依据的预测。作者预计,新实验数据的引入及其后续整合将进一步增强RENAISSANCE的预测能力。
讨论
代谢在决定生物整体健康中起着关键作用,异常代谢与多种疾病有关。深入理解代谢过程对于药物开发、个性化治疗和生物技术进步至关重要。动力学模型提供了最详尽的代谢数学表示,但传统构建方法复杂且耗时。RENAISSANCE框架利用机器学习和自然进化策略,无需训练数据即可生成动力学模型,大大提高了构建效率。它可以在普通工作站上快速训练生成器,生成大量模型,并适应多种生物化学属性和生理条件。RENAISSANCE的开源代码将有助于实验人员和建模者应用该框架并整合多种数据。
编译 | 于洲
审稿 | 王梓旭
参考资料
Choudhury S, Narayanan B, Moret M, et al. Generative machine learning produces kinetic models that accurately characterize intracellular metabolic states[J]. Nature Catalysis, 2024: 1-13.
内容中包含的图片若涉及版权问题,请及时与我们联系删除

评论
沙发等你来抢