
DRUGAI
今天为大家介绍的是来自中山大学杨跃东团队的一篇论文。酶在众多生物过程中起着至关重要的作用,其中EC编号是定义酶功能的常用方法。然而,现有的EC编号预测技术并未充分认识到酶活性位点和结构特征的重要性。为此,作者提出了一种基于几何图学习的EC编号预测器——GraphEC,它使用ESMFold预测的结构和预训练的蛋白质语言模型。具体来说,作者首先构建了一个模型来预测酶的活性位点,并利用该模型来预测EC编号。通过引入同源信息的标签扩散算法(label diffusion algorithm),进一步提升了预测的准确性。同时,模型还预测了酶的最佳pH值,以反映酶催化反应的特性。实验表明,该模型在活性位点、EC编号及最佳pH值的预测方面,相较于其他先进方法具有更优表现。进一步的分析揭示,GraphEC能够从蛋白质结构中提取功能信息,强调了几何图学习的有效性。该技术可用于识别未注释的酶功能,并预测其活性位点和最佳pH值,具有推动合成生物学、基因组学等领域研究的潜力。

酶通过催化多种反应在各类生物过程中起着重要作用。识别酶的功能对代谢和疾病的研究具有重要意义。EC编号以四位数字的结构形式表达酶的功能,为酶工程领域提供统一的体系。然而,EC编号的实验测定过程耗时且成本高昂。因此,发展用于识别EC编号的计算方法变得尤为必要。
计算方法可以分为基于同源性、基于结构和基于机器学习的方法。基于同源性的方法假设高度相似的酶具有相似的功能,并利用比对工具对酶功能进行注释。这些方法主要依赖于序列相似性,但在缺乏相似序列时,其覆盖率有限。为提高覆盖率,基于结构的方法通过扫描结构相似的蛋白模板来识别一致的功能。例如,COFACTOR通过将查询结构与BioLiP库中已知结构和功能的蛋白质进行比较,以实现功能注释。尽管这些方法有所改进,但由于缺乏高质量模板,仍面临挑战。为了跳出相似序列和模板的限制,基于机器学习的方法应运而生。早期的机器学习方法先提取重要特征,再利用机器学习算法来识别相应的EC编号。然而,这些算法的性能高度依赖于人工设计的特征,难以适应不断扩展的酶序列。
近年来,深度学习方法在酶功能注释方面取得了成功。为避免人工特征提取,DEEPre利用CNN和RNN组件来捕捉卷积和序列特征。ProteInfer使用空洞(dilated)卷积网络来建立蛋白质空间与酶功能空间之间的映射。GrAPFI使用InterPro签名作为领域信息,在加权无向图上进行标签传播(label propagation)。CLEAN是另一种深度学习方法,通过对比学习获取丰富的嵌入特征,在EC编号识别中实现了更高的准确性和更广的EC覆盖范围。然而,这些方法仍存在两个局限性:第一,它们仅使用蛋白质序列而未结合蛋白质结构,从而失去了结构中隐含的重要特征;第二,它们未在酶功能分析中利用酶活性位点的关键信息。
由于缺乏天然结构,现有方法未能充分利用蛋白质结构中的信息。Lin等人提出了预训练语言模型ESMFold,用于精确且快速的结构预测,其准确性与AlphaFold2相当,但推理时间缩短了高达60倍。在ESMFold预测结构的帮助下,几何图学习可以高效提取结构信息,这一技术已在蛋白质设计和对接中证明了其优势。为了增强几何图学习,一些研究尝试使用无监督语言模型(如ProtTrans和ESM-1b)结合有用的序列嵌入信息。
另一方面,酶的活性位点通常位于酶的表面,在催化反应或结合底物中发挥重要作用。这些位点在进化过程中高度保守,对酶功能具有决定性影响。因此,在分配EC编号时,酶的活性位点意义很大。然而,当前用于预测酶活性位点的方法主要依赖模板或手工设计的特征,无法跟上迅速增长的数据,这凸显了对快速且准确的酶活性位点预测工具的需求。除了活性位点外,标签扩散算法也被开发用于蛋白质功能预测,可以转移功能相关的数据,辅助识别EC编号。
模型部分
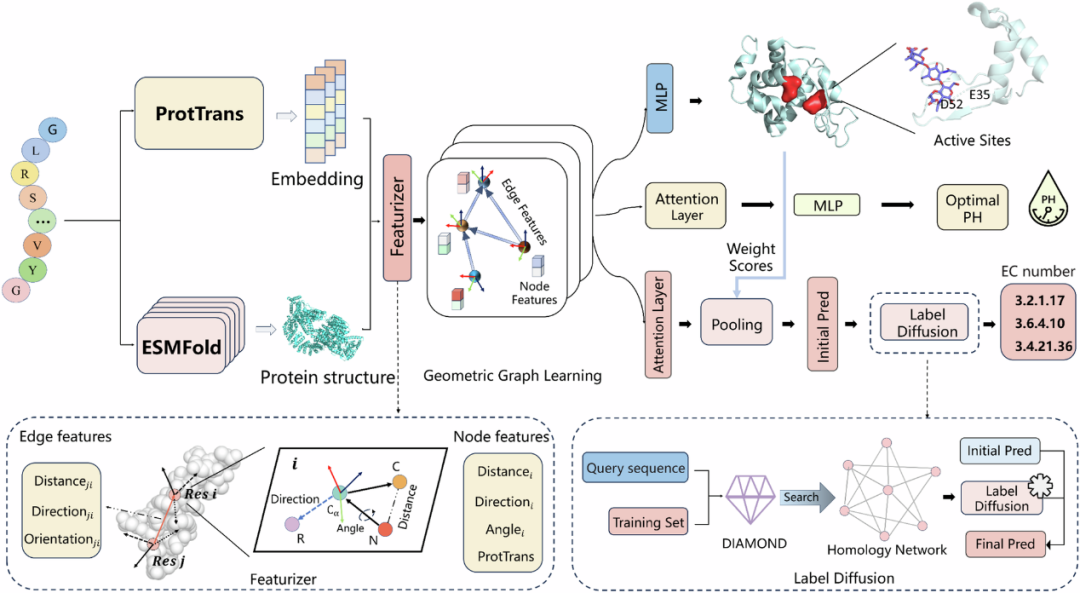
图 1
GraphEC是一种基于几何图学习的精准EC编号预测器,将酶的活性位点和预测的蛋白质结构整合到酶功能预测中(图1)。给定一个蛋白质序列,其结构由ESMFold预测并用于构建蛋白质图。几何特征通过预测结构提取,并通过预训练语言模型ProtTrans计算的序列嵌入进行增强。这些特征输入到几何图学习网络中,用于学习几何嵌入,并应用于活性位点、EC编号和最佳pH值的预测。GraphEC-AS首先预测酶的活性位点,并为每个残基分配权重分数。在这些权重分数的指导下,初步的EC编号预测通过注意力层和池化层计算得出,并通过提取同源信息的标签扩散算法进一步优化。最后,模型通过注意力池化扩展到最佳pH值的预测,以更好地表示反应条件(GraphEC-pH)。
酶活性位点预测
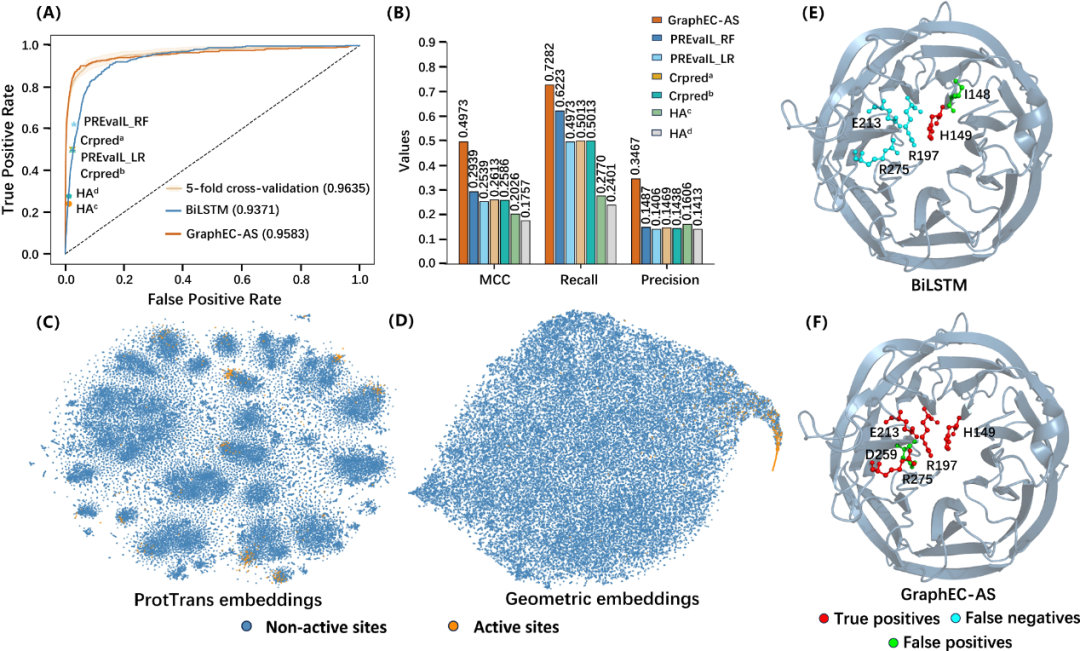
图 2
作者首先评估了GraphEC-AS在独立测试集TS124上的残基级酶活性位点预测。图2A显示,GraphEC-AS在五折交叉验证中的AUC(ROC曲线下的面积)为0.9635,在TS124上的AUC为0.9583,表明该模型具有良好的稳健性。六种对比方法(PREvaIL_RF、PREvaIL_LR、CRpred(带坐标的残基)、CRpred(所有残基)、HA(残基身份筛选)、HA(组合筛选))的ROC曲线位于GraphEC-AS和BiLSTM(不包含结构信息的模型)之间,凸显了几何信息的重要性。
在MCC(Matthews相关系数)、召回率和精准度方面(图2B),GraphEC-AS始终表现最佳。排名第二的PREvaIL_RF分别取得了0.2939、0.6223和0.1487,较GraphEC-AS低40.9%、14.5%和57.1%。此外,GraphEC-AS在TS124上的F1得分为0.4698,而第二名的PREvaIL_RF得分为0.240,相比GraphEC-AS下降了48.9%。PREvaIL方法需要使用PSI-BLAST计算耗时的进化特征,而GraphEC-AS可以快速且准确地识别酶活性位点。
GraphEC-AS在TS124上的已学习特征结果上进一步展示了其优势。图2C中的ProtTrans嵌入分布较为分散,而GraphEC-AS学习的几何嵌入(图2D)则清晰地区分了活性位点和非活性位点。这表明几何图学习能够有效识别它们之间的关键差异。作者进一步使用TM-align评估了ESMFold预测结构质量对TS124的影响。超过85%的蛋白质TM-Score高于0.8,反映了ESMFold预测结构的高质量。AUC值随着TM-Score的增加而提升,这说明了高质量预测结构的重要性,并强调了使用ProtTrans增强特征嵌入的必要性。
图2E和图2F对比了BiLSTM和GraphEC-AS对同一个例子(顺式粘康酸环化酶)预测的三维结构。GraphEC-AS识别了所有四个活性位点,而BiLSTM仅检测到H149,因为其缺乏局部结构特征的识别能力。相比H149,其余活性位点在序列上距离较远(超过20个残基),但在结构上却相距较近(小于16 Å)。这些结果表明,GraphEC-AS能够学习局部结构信息。
酶EC编号分类
在预测的活性位点指导下,GraphEC用于识别酶的EC编号。GraphEC在两个独立测试集NEW-392和Price-149上进行了评估,其中NEW-392包含392个酶序列,涵盖177个不同的EC编号,而Price-149是Price等人验证的实验数据集。与四种先进的EC编号预测器(包括CLEAN、ProteInfer、DeepEC、ECPred、GrAPFI和ECPICK)相比,GraphEC在多项指标上表现更佳。
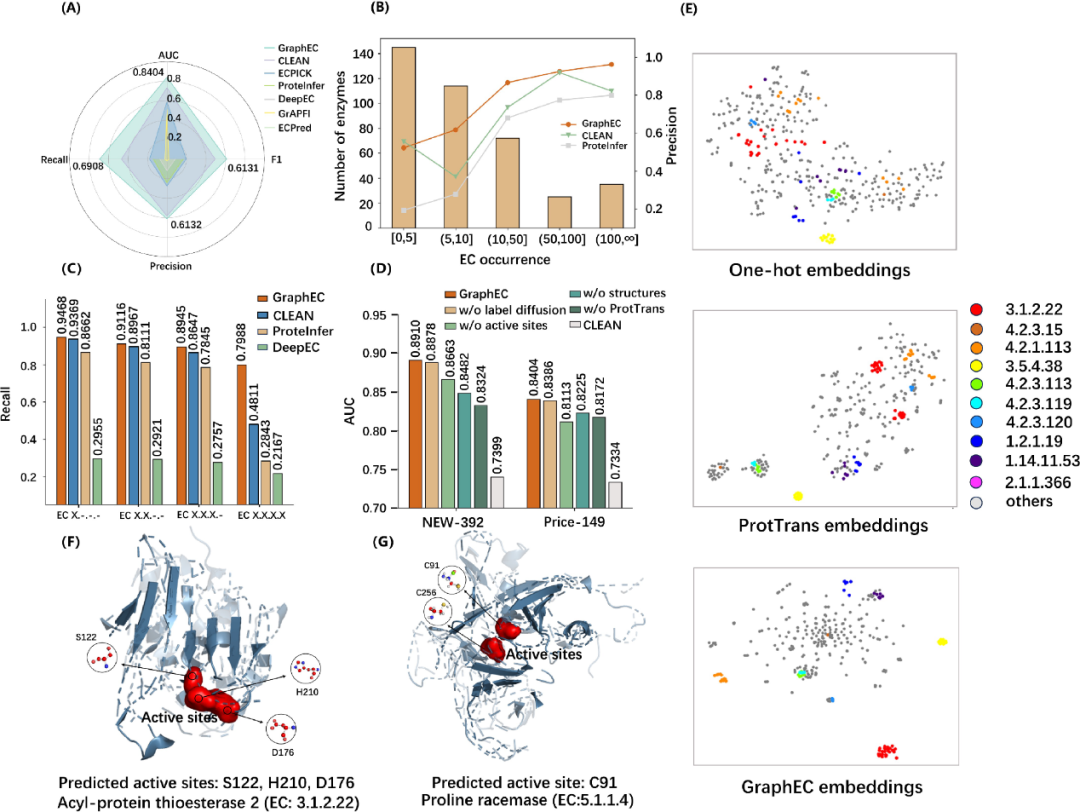
图 3
图3A显示,GraphEC在Price-149上的AUC、召回率(recall)、精准度(precision)和F1分别为0.8404、0.6908、0.6132和0.6131,分别比表现第二好的CLEAN高出14.6%、47.9%、4.9%和23.9%。在NEW-392上,GraphEC在AUC(0.8910)、召回率(0.7988)和F1(0.5910)方面均取得最佳值。
GraphEC还在不同层级的EC编号和各EC编号在训练集中的出现频率上进行了评估。考虑到训练集中EC编号频率可能对模型性能的影响,作者在NEW-392上根据EC编号在训练集中出现的次数评估了精度(图3B)。超过66.0%的酶出现次数少于十次,只有8.9%的酶出现次数超过100次,显示出数据集的挑战性。正如预期,预测低频EC编号较为困难。然而,GraphEC在不同出现频率的EC编号上始终表现出较高的精度,突显了模型的优越性。
EC编号的四个数字分别对应酶功能分类的不同层级,第一至第四位数字代表逐级分解的功能层级。在NEW-392上,GraphEC相较于CLEAN的召回率分别在第一至第四层级提升了1.1%、1.7%、3.4%和66.0%,其召回率分别为0.9468、0.9116、0.8945和0.7988(图3C)。随着层级的增加,GraphEC的优势变得更加明显,表明了模型的有效性。
在EC编号预测中考虑活性位点的应用,作者评估了活性位点突变的影响。首先,作者基于预测结果(得分> 0.5)识别了NEW-392和Price-149中的酶活性位点。随后,将这些活性位点突变为丙氨酸(A),并比较了突变前后真实EC编号的预测得分。突变后,真实EC编号的预测得分有所下降,这表明活性位点的突变对EC编号预测有影响。在突变的酶中,59.1%被识别为非酶类,如L-2-羟基戊二酸脱氢酶(Uniprot ID: A0A011QK89)和法呢基焦磷酸合酶(Uniprot ID: B4YA15)。此外,作者比较了突变前后活性位点的预测得分,发现突变后活性位点的预测得分下降,这表明模型对突变后活性位点的关注度降低。
消融实验分析
作者对GraphEC进行了模块消融研究,以评估各模块的贡献。当移除标签扩散时,AUC值略有下降(图3D),这可能是因为GraphEC能够学习同源信息的能力。当移除活性位点指导时,NEW-392和Price-149的AUC分别下降了2.8%和3.5%,这表明活性位点指导的重要性。为评估ESMFold预测结构的影响,构建了一个不考虑几何信息的基线模型(BiLSTM)。在没有结构信息的情况下,NEW-392和Price-149的AUC分别下降了4.8%和2.1%,表明预测结构至关重要。ProtTrans嵌入用于增强节点特征,移除后AUC分别下降了6.6%和2.8%。这里使用的ProtTrans嵌入是残基级别的表示,与CLEAN中使用的蛋白质级别ESM-1b表示(均值表示)不同。
如图3E所示,作者将学习到的几何嵌入(GraphEC嵌入)与ProtTrans嵌入和one-hot嵌入在NEW-392上进行了比较。在十个最常见的EC编号中,one-hot嵌入的区分能力有限。ProtTrans嵌入能够大致区分这些EC编号,但无法将3.1.2.22和4.2.1.113所属的类别聚类。而GraphEC嵌入则可以清晰地分离这些EC编号,展示了其对不同EC编号的强大表达能力。同样地,在Price-149数据集中,one-hot嵌入缺乏区分能力,ProtTrans嵌入提供了基本的区分,而GraphEC嵌入则能进一步区分这些编号。
为评估预测结构的重要性,作者将ESMFold预测的结构替换为AlphaFold2预测的结构。在NEW-392上,使用AlphaFold2预测的结构时,AUC、召回率、精准度和F1分别为0.9004、0.8267、0.5745和0.6044(补充表S6),略高于使用ESMFold预测结构时的表现。在Price-149上,使用AlphaFold2预测结构和ESMFold预测结构时,性能相当。结果表明,ESMFold可以在比AlphaFold2更短的时间内生成具有相近准确度的结构。
GraphEC能够捕捉酶的功能区域
为验证GraphEC是否可以识别功能区域,作者研究了预测的酶活性位点、多头注意力分数与真实活性位点之间的关系。如图3F所示,酰基蛋白硫酯酶2的真实活性位点为S122、D176和H210,这些位点通过GraphEC-AS正确预测并用于指导EC编号预测。多头注意力分数在接近真实活性位点时趋于更高,表明模型能够关注到功能区域。类似地,脯氨酸消旋酶的酶活性位点也被准确识别,多头注意力分数在接近真实活性位点时尤为突出(图3G)。这些结果表明,GraphEC能够捕捉酶的功能区域。
酶最适PH的预测
由于酶的pH值对酶功能至关重要,作者还纳入了酶的最佳pH值预测。为训练模型,作者从Brenda数据库(2023年1月发布)中整理了一个新数据集,包含4110个序列相似性小于25%的蛋白质。数据集按照4:1的比例根据提交时间分为训练集(Brenda-train,3297个酶)和独立测试集(Brenda-test,813个酶)。
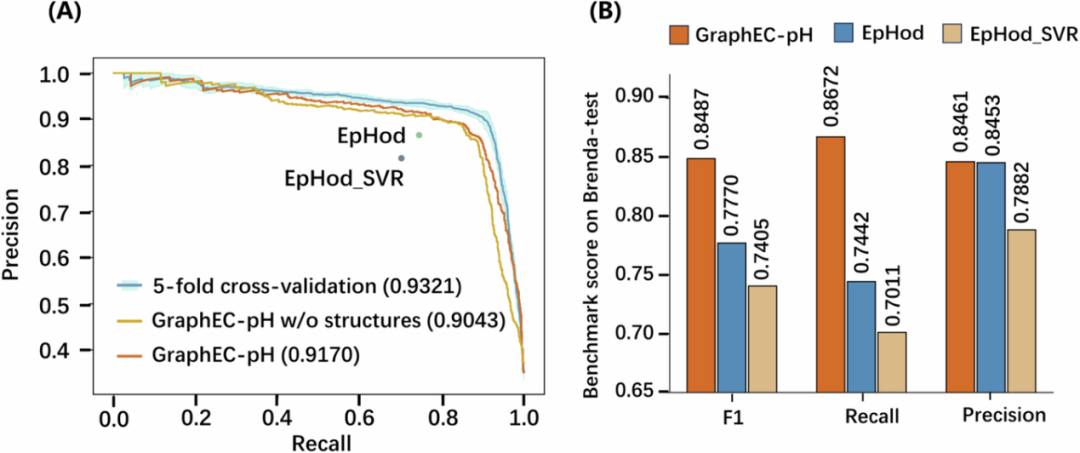
图 4
如图4A所示,GraphEC-pH在五折交叉验证中的AUPR(精确率-召回率曲线下的面积)为0.9321,在测试集上的AUPR为0.9170,显示了模型的稳健性。当移除结构信息时,GraphEC-pH的AUPR下降了1.4%。相比之下,最新的两种方法EpHod和EpHod_SVR表现较差,位于GraphEC-pH的精确率-召回率曲线下方。相应地,GraphEC-pH的F1、召回率和精准度分别为0.8487、0.8672和0.8461,分别比第二好的方法EpHod高出9.2%、16.5%和0.09%(图4B)。这些结果展示了模型的优越性能。
随后,作者评估了模型区分Brenda测试集中由DIAMOND搜索的289对同源酶对之间差异的能力。在这些同源酶对中,超过87.9%(254对)具有相同的最佳pH类型(即“酸性”-“酸性”和“非酸性”-“非酸性”),GraphEC-pH能够正确识别其中的95.7%(243对)。只有35对酶表现出不同的最佳pH类型(即“酸性”-“非酸性”),其中GraphEC-pH正确区分了14对,比EpHod多出75%(EpHod正确区分8对)。这些结果表明,GraphEC-pH在一定程度上能够辨别同源酶之间的差异。
GraphEC能从酶结构中学习功能信息
为发现新的酶功能,作者从Swiss-Prot(2024年1月发布)收集了共计570,830个蛋白序列。经过去除序列相似性大于25%的蛋白质以及与训练数据集相似性超过25%的蛋白质后,剩下52,037个没有EC编号注释的蛋白质。这些蛋白质通过GraphEC和CLEAN进行了注释,其中超过21%的蛋白质包括相同的EC编号注释。对于每个蛋白质,获取了预测的EC编号,并计算了与训练集中相同EC编号蛋白质的TM-Score,随后使用这些蛋白质的最大TM-Score进行分析。GraphEC总体得分较高,超过82%的蛋白质通过Foldseek显示出比CLEAN更高的TM-Score。
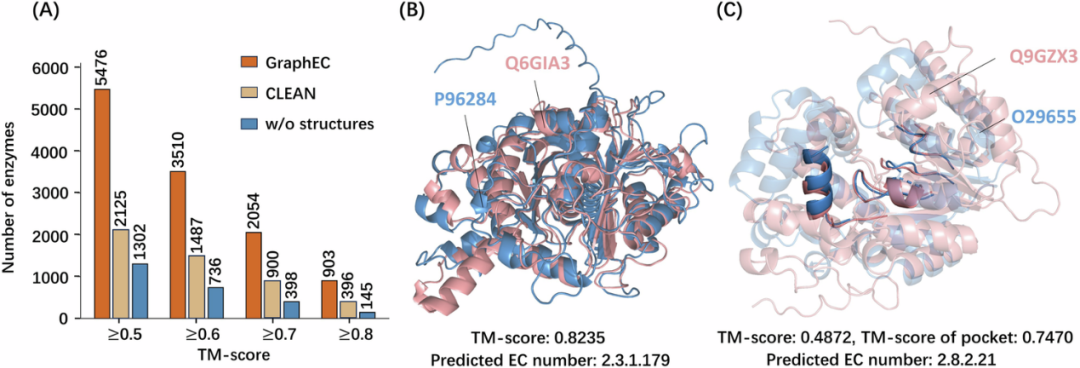
图 5
在比较最大TM-Score超过不同阈值的酶数量时(图5A),GraphEC在0.5、0.7、0.8和0.9阈值下分别比CLEAN多出158%、136%、128%和128%。与CLEAN相比,GraphEC发现的最大TM-Score超过0.8的新酶功能部分列于补充数据集1。尽管序列相似性较低,GraphEC能够从结构相似度高的酶中学习功能信息(图5B)。即使TM-Score较低,酶活性位点周围的酶口袋仍能对齐(图5C),显示了GraphEC从酶结构中学习关键功能信息的能力。
参考资料
Song, Y., Yuan, Q., Chen, S., Zeng, Y., Zhao, H., & Yang, Y. (2024). Accurately predicting enzyme functions through geometric graph learning on ESMFold-predicted structures. Nature Communications, 15(1), 8180.
内容中包含的图片若涉及版权问题,请及时与我们联系删除

评论
沙发等你来抢