
DRUGAI
今天为大家介绍的是来自唐建团队的一篇论文。提高抗体对靶抗原的结合亲和力是抗体开发中的关键任务。这篇文章提出了一个可预训练的几何图神经网络GearBind,并探索了该网络在亲和力成熟(affinity maturation)中的应用潜力。GearBind有着以下三个特性(1)构建多关系图(2)多层次的几何消息传递(3)在大量无监督蛋白结构数据上进行对比学习预训练,使得它在SKEMPI和独立测试集上的模型性能优于先前的sota方法。基于GearBind,作者构建了一个强力的集成模型,成功地增加了两个抗体的结合,即使这两个抗体的格式与靶抗原截然不同。设计的抗体突变体的ELISA EC50值最多降低17倍,KD值最多降低了6.1倍。这些好的实验结果显示了几何深度学习与预训练在大分子反应建模任务中的用处。

抗体在人体免疫系统中起着重要作用,并因其能够选择性且高特异性地结合靶抗原而成为强大的诊断和治疗工具。在体内,抗体会经历亲和力成熟,通过体细胞高频突变和克隆选择,使结合靶标的亲和力逐渐增强。当出现新的抗原时,重新利用已知抗体,或者从天然、新设计的抗体库筛选出的治疗性抗体,往往需要进行体外亲和力成熟,以提高其结合亲和力到理想的水平,通常是亚纳摩尔级。
体外抗体亲和力成熟的湿实验方法通常涉及构建突变库并通过展示技术进行筛选。这些方法虽然在过去几年中有了显著改进,但整体上依然耗时耗力且成本高昂,通常需要2-3个月或更长时间才能完成。考虑到可能突变的组合搜索空间,抗体的互补决定区(complementarity-determining region,CDR)上通常有50-60个氨基酸残基,这些区域在体内具有高度变异性,并贡献了大部分结合自由能ΔGbind。以往的研究显示,成功的亲和力成熟往往需要多次点突变。对抗体CDR区域中成千上万种可能的点突变组合(60个残基 × 每个残基19种变异)进行实验是非常困难的,甚至是不可能的。因此,迫切需要一种快速且准确的计算方法来缩小搜索空间。
然而,计算亲和力成熟方法要在速度和准确性之间取得平衡并非易事。基于经验力场的分子动力学方法依赖于人工知识和抽象来评估突变后的结合自由能变化。然而,精确的模型通常速度过慢,无法用于对成千上万种突变(更不用说它们的组合)进行排序。近年来,许多机器学习方法将亲和力成熟问题表述为基于结构的结合自由能变化的预测问题。然而,尽管蛋白质侧链构象对蛋白质-蛋白质相互作用至关重要,大多数现有方法对原子级几何信息的建模方式是间接的或不完整的,如使用手工特征或残基级特征。这些方法无法充分解决侧链原子之间的复杂相互作用。另一个关键问题是机器学习模型需要大量的配对结合亲和力数据,以确保其准确性和可靠性。据作者所知,当前最大的公开蛋白质-蛋白质结合自由能变化数据集SKEMPI v2.0,仅包含348个蛋白质复合物上的7085个ΔΔGbind测量值,这一数据量相比于像AlphaFold2和ESM2等基础蛋白质模型的训练集规模显得微不足道。
模型部分
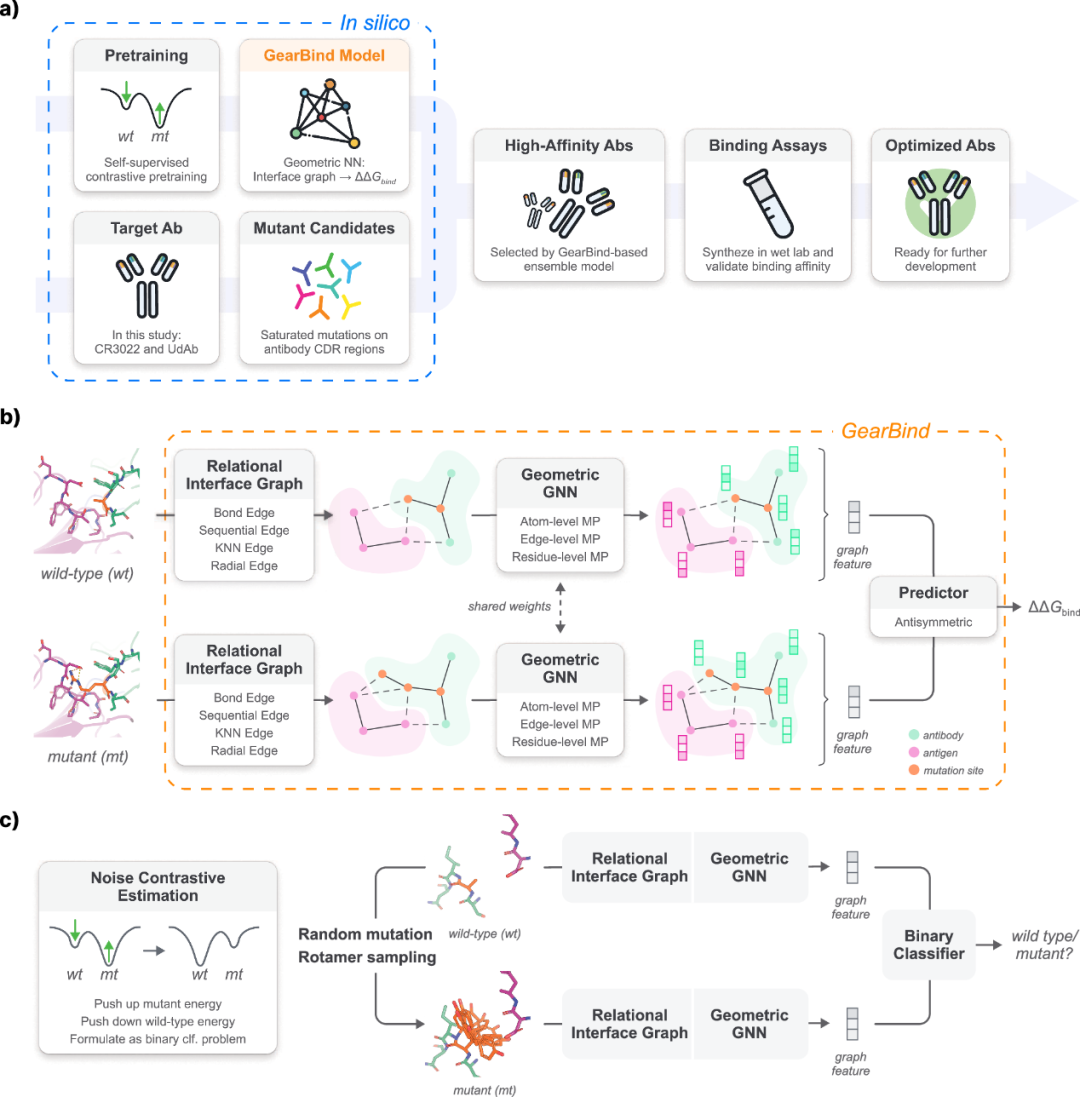
图 1
作者引入了GearBind,这是一种可预训练的深度神经网络,利用多层次几何信息传递来模拟复杂的蛋白质-蛋白质相互作用。作者使用对比预训练技术,在大规模蛋白质结构数据集上进行训练,以将重要的结构信息融入模型中(图1)。
具体地说,当一个蛋白质复合物结构输入到GearBind时,首先会对于接触界面构建一个多关系的界面原子图来捕捉(舍弃掉其他原子),而图中四种关系(边)分别为:Bond Edge,Sequential Edge,KNN Edge,Radial Edge。其中,如果两个原子在氨基酸序列中的相对距离小于3,那它们之间就形成一条sequential edge,而sequential edge边种类有5种(-2,-1,0,1,2),是通过节点间的距离确定种类的;如果两个原子空间上距离小于5A,则形成一条radial edge;而每个原子与空间上最近的10个邻居形成KNN edge,保证图连通性。
随后,通过在界面图上应用几何关系图神经网络GearNet,获得原子层的表示。在此基础上,为了建模边之间的反应,作者将原子图中的每条边视为一种节点,叫“线节点”(line node),而如果原子图中两个边有公共顶点,两个“线节点”就会形成一条“线边”(line edge)(线边特征为角度信息),从而构建出“线图”(line graph)。通过在该线图上进行信息传递,捕捉边级别的相互作用,类似于AlphaFold三角注意力的稀疏版本。
最后,在聚合每个残基的原子和边级别表示后,应用几何图注意力层在残基之间传递信息。这种多层次的信息传递方案将多粒度的结构信息注入到学习到的表示中,使其在ΔΔGbind预测任务中非常有用。
虽然GearBind可以在标注的ΔΔGbind数据集上从零开始训练,但如果训练数据量有限,可能会出现过拟合或泛化能力差的问题。为了解决这个问题,作者提出了一种自监督预训练任务,利用CATH数据库中的大规模无标签蛋白质结构,模型通过对比原生结构与随机突变结构(突变结构的侧链扭转角从构象体库中采样)进行训练,提升突变体能量,降低野生型能量,将其建模为一个二分类问题(图1c)。
模型在SKEMPI数据集上的性能评估
作者通过在SKEMPI v2.0上进行了五折交叉验证(按复合物划分),验证了GearBind的性能。作者的划分策略规定,每个测试集与相应的训练集没有共同的PDB复合物,这比按突变划分更现实,因为按突变划分的策略中,测试集中的野生型蛋白质复合物甚至突变位点可能在训练中出现。作者将GearBind和GearBind+P(经过预训练并在SKEMPI上微调的GearBind)与先进的物理学工具FoldX、Flex-ddG,以及深度学习方法Bind-ddG进行了比较。
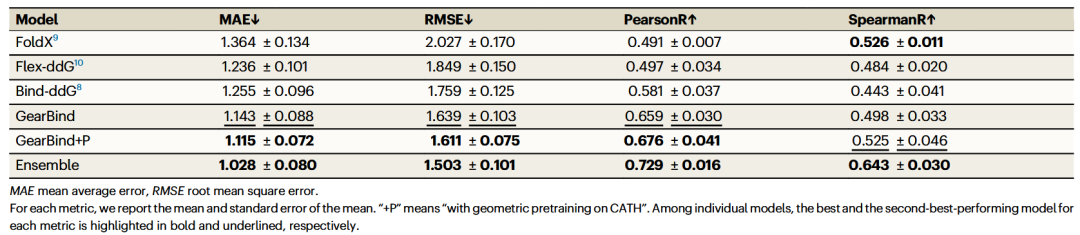
表 1
结果(表1)显示,GearBind凭借其多关系图构建和多层次信息传递方案,在平均绝对误差(MAE)、均方根误差(RMSE)和Pearson相关系数方面优于基线模型,而在Spearman相关系数上仅次于FoldX。对GearBind进行预训练进一步提升了性能,Spearman相关系数提升了5.4%,Pearson相关系数提升了2.6%,MAE减少了2.4%,RMSE减少了1.7%。这突出显示了从大规模、无标签蛋白质结构数据中进行有效知识转移的优势。
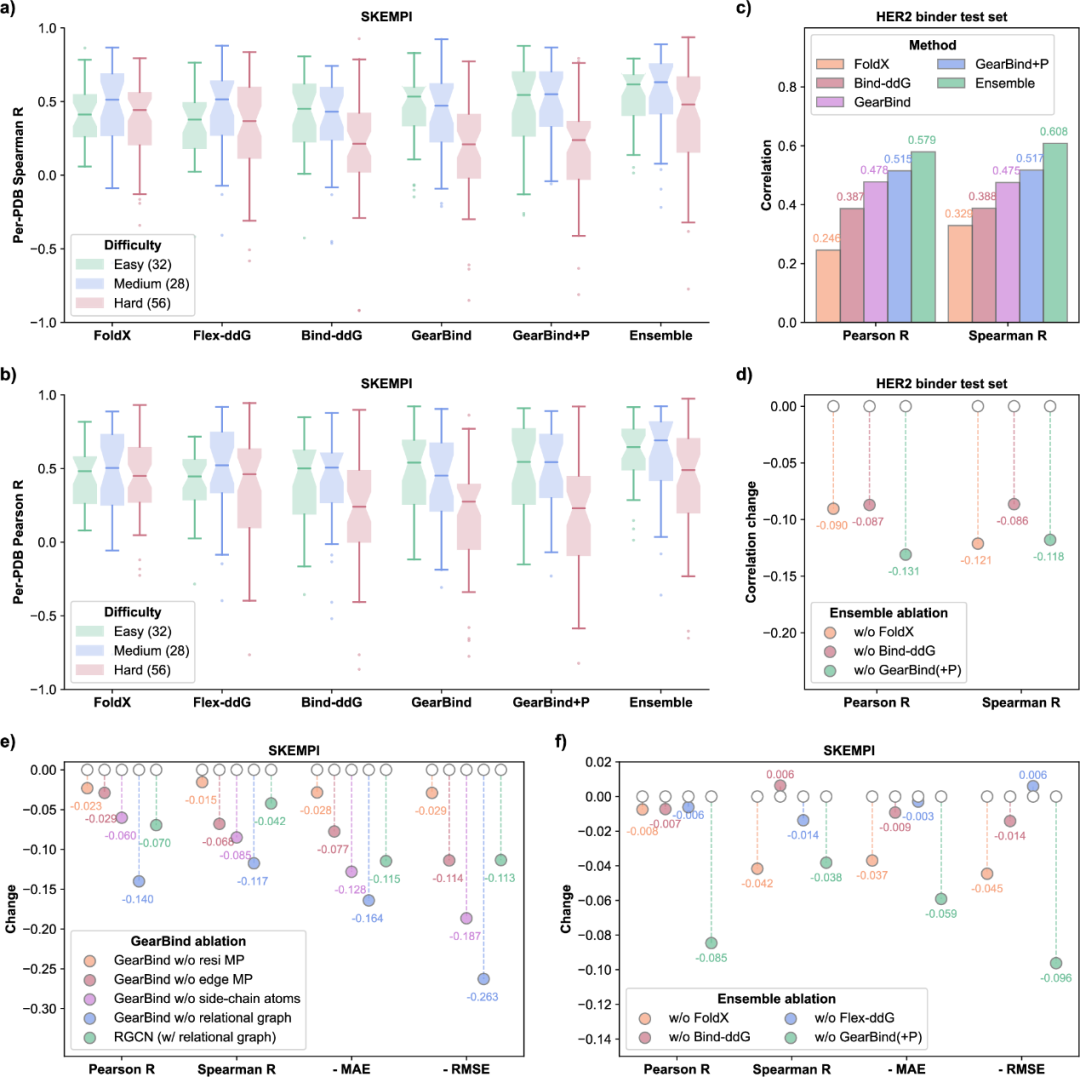
图 2
为了了解GearBind中关键架构设计选择的贡献,作者在SKEMPI上对五个GearBind变体进行了性能基准测试,这五个变体分别排除了GearBind的一些成分(如关系边,残基层)。如图2e所示,所有测试的GearBind变体在四个指标上的表现都不如GearBind。从GearBind中排除边和残基层的信息传递导致Spearman相关系数分别下降了13%和3%,这突显了在特征提取过程中结合多层次信息的优势。从界面图中排除侧链原子使性能进一步下降(Spearman相关系数下降15%),说明明确建模全原子结构的重要性。值得注意的是,用KNN图替换多关系界面图导致Spearman相关系数大幅下降23%,而在多关系图上训练简单的RGCN模型的性能与Bind-ddG相当(相比GearBind,Spearman相关系数下降9%,但相比Bind-ddG提高了2%)。这些结果表明,多关系图构建策略是GearBind的关键组成部分。
基于GearBind的集成模型用于计算机亲和力成熟
为了了解基准模型在SKEMPI上的表现,作者根据目标难度将SKEMPI数据集分组,并绘制了每个模型在不同难度目标上的Pearson相关系数和Spearman相关系数。根据训练集中具有较高结构相似性(TM分数>0.8)的数据点数量,SKEMPI中的PDB id被分类为“简单”(训练集中有50个以上相似数据点)、“中等”(1-50个)和“困难”(0个)。深度学习模型如Bind-ddG、GearBind和GearBind+P在简单目标上表现优于基于物理的方法如FoldX和Flex-ddG,但在困难目标上情况发生逆转(图2a、b),显示出在泛化能力方面仍有提升空间。作者还研究了引起结合自由能绝对变化为低(<0.5 kcal/mol)、中等(0.5-2)、高(>2)突变的性能。当结合水平变化更剧烈时,所有模型表现更好。GearBind在这一区域的表现尤为突出,其Pearson相关系数为0.707,而FoldX为0.411,显示出其识别显著增强或破坏结合的突变的潜力。当∣ΔΔGbind∣较小时,所有方法的预测与实验ΔΔGbind值的相关性都很低,这暗示可能是数据噪声或当前工具在建模较弱且更复杂相互作用时存在不足。
为结合基于物理和深度学习方法的优势,作者使用所有基准模型的集成进行后续的计算亲和力成熟。集成模型的预测值是FoldX、Flex-ddG、GearBind、GearBind+P和Bind-ddG预测值的简单平均。结果显示,提出的集成模型在所有四个评估指标上均优于单个模型(表1)。作者通过排除各个模型并在SKEMPI上评估性能,分析了每个模型对集成的贡献。结果(图2f)显示,排除GearBind和GearBind+P对整体性能的影响最大。具体而言,在Pearson相关系数指标上,单独排除FoldX、Flex-ddG和Bind-ddG导致的下降微乎其微(小于0.01),但移除GearBind则导致显著下降(超过0.08)。作者还注意到,尽管FoldX单独使用时并非表现最佳的模型,但将其从集成中移除却引起Spearman相关系数最大幅度的下降。这表明FoldX在补充深度学习模型并构建稳健、精确的集成模型中发挥了重要作用。
在HER2结合体测试集上的评估
在建立并训练了模型(基于SKEMPI)后,作者测试了它们在HER2结合物测试集上的性能,这些数据集来自Shanehsazzadeh等人。该数据集包含通过表面等离子体共振(SPR)测量的高质量结合亲和力数据,共有419个新设计的CDR环的HER2结合物。数据集中的抗体是Trastuzumab的变体,具有较高的编辑距离(平均7.6),这对训练于低编辑距离数据的ΔΔGbind预测器来说可能是一个挑战。在基准方法中,GearBind+P在Pearson相关系数和Spearman相关系数上表现最佳(图2c)。随后,作者将所有基准模型的预测值取平均,形成集成模型,并逐一排除各个模型以衡量性能变化。结果显示,排除GearBind(+P)对Pearson相关系数的影响最大,而排除FoldX对Spearman相关系数的影响最大,GearBind(+P)紧随其后(图2d)。
CR3022和anti-5T4 UdAb的亲和力成熟分析
为验证方法的有效性,作者选择了两种抗体CR3022和anti-5T4 UdAb作为亲和力成熟的研究对象。CR3022抗体最初从一位康复的SARS患者中分离出,后来发现其可以结合SARS-CoV-2。而针对癌胎抗原5T4的UdAb以其卓越的稳定性著称。这两种抗体具有不同的格式并靶向不同的抗原。两种抗原在SKEMPI中各只有一个结构上相似的蛋白质链(TM分数>0.8),且结合位点不同,使它们成为对本研究流程的挑战性目标。
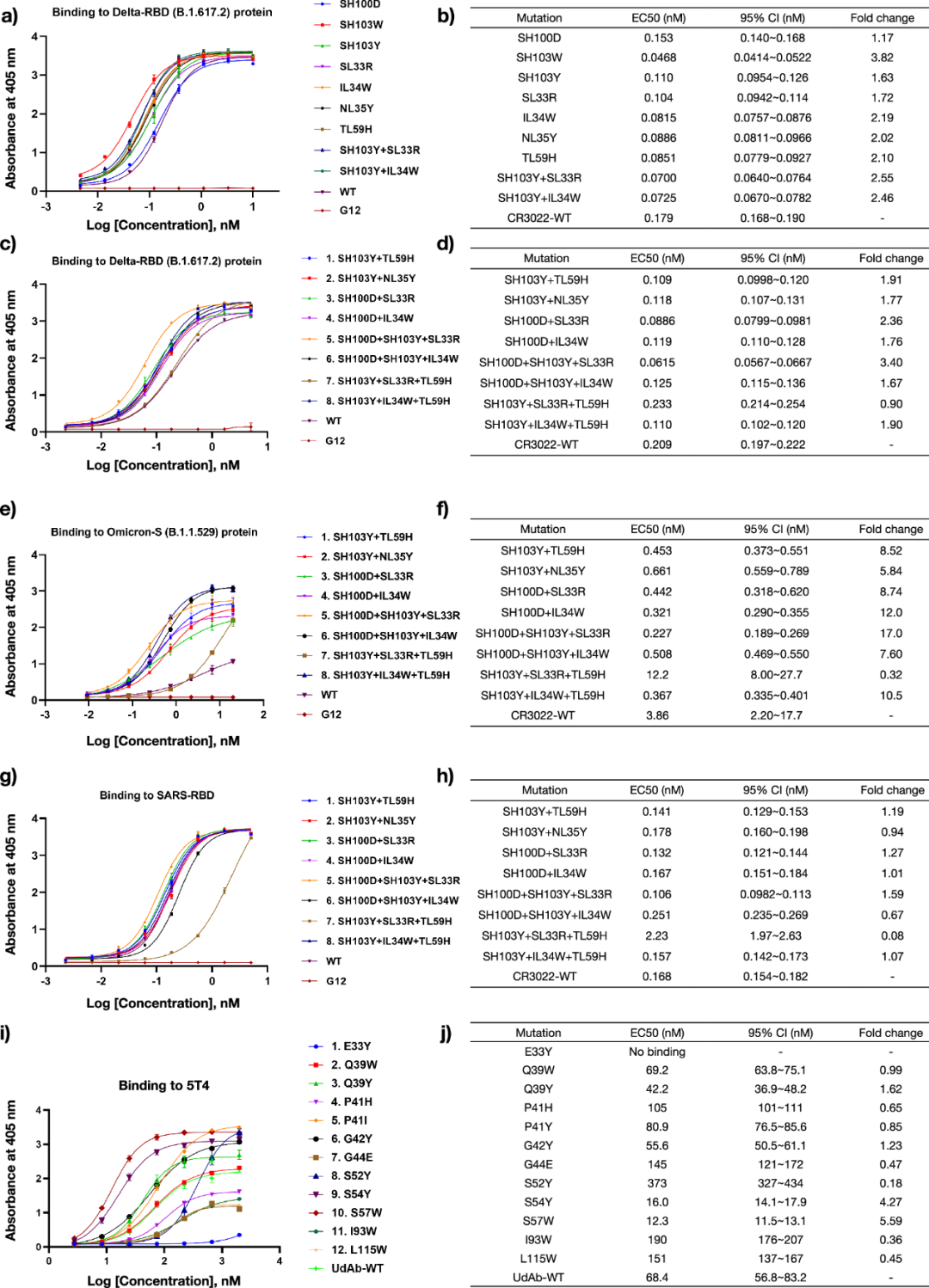
图 3
对于CR3022,作者根据集成模型对野生型、BA.1.1和BA.4 SARS-CoV-2毒株的RBD亲和力变化的预测,在第一轮实验验证中选出了12个突变体。值得注意的是,野生型和Delta RBD在与CR3022的界面处具有相同的氨基酸。在ELISA预实验中,作者测试了这些突变体与SARS-CoV-2 Delta毒株RBD的结合情况,抗原浓度为100 nM。12个候选突变体中有9个显示出比野生型CR3022更强的结合力。在抗原浓度降低至10 nM的进一步验证中,这9个候选突变体的EC50值均低于野生型CR3022(图3a、b)。基于这些结果,作者将表现良好的CR3022突变结合起来,设计并合成了8个含有双重或三重突变的候选体作为第二轮设计。8个多点突变体中有7个对Delta RBD的结合力增强,ELISA EC50值比野生型低1.8–3.4倍。其中三重突变体SH100D+SH103Y+SL33R的EC50最低,为0.06 nM(图3c、d)。针对奥密克戎刺突蛋白,这7个多点突变体的结合力再次提高了7.6–17.0倍,EC50值均为亚纳摩尔级,三重突变体SH100D+SH103Y+SL33R仍表现最佳(图3e、f)。接下来,作者测试了第二轮设计的突变体对SARS-CoV RBD的结合力,以评估CR3022针对SARS-CoV-2 RBD的结合优化是否会对其原始靶标产生显著变化。8个突变体中有7个在ELISA EC50方面对SARS-CoV RBD未表现出显著变化(图3g、h)。综上所述,这些结果证明了基于GearBind的流程在CR3022抗体亲和力优化中的成功。
编译 | 黄海涛
审稿 | 王梓旭
参考资料
Cai, H., Zhang, Z., Wang, M., Zhong, B., Li, Q., Zhong, Y., ... & Tang, J. (2024). Pretrainable geometric graph neural network for antibody affinity maturation. Nature Communications, 15(1), 7785.
内容中包含的图片若涉及版权问题,请及时与我们联系删除

评论
沙发等你来抢