
DRUGAI
今天为大家介绍的是来自美国西奈山伊坎医学院的Ron Do教授团队的一篇论文。识别慢性疾病的遗传驱动因素对于药物发现至关重要。在本文中,作者开发了一种机器学习辅助的遗传优先评分(ML-GPS),它将遗传关联与预测的疾病表型结合起来,以增强靶点发现。首先,作者构建了梯度提升模型,用于预测英国生物样本库中112种慢性疾病的表型代码,并分析预测表型和观察表型与常见、罕见和极罕见变异的关联,以建立等位基因系列模型。作者通过带有连续特征编码的梯度提升算法,将这些关联与现有证据整合,构建了ML-GPS,并在Open Targets平台上训练其药物指示预测能力,并在SIDER数据库中进行外部测试。随后,作者生成了ML-GPS对2,362,636个基因-表型代码对的预测结果。作者发现,使用预测表型能比观察表型识别出更多跨等位基因频谱的遗传关联,从而显著提升了ML-GPS的性能。ML-GPS扩展了药物靶点的覆盖范围,其前1%的评分为15,077个先前无支持的基因-表型代码对提供了支持。ML-GPS还能够识别已知的靶点-疾病关系、尚无指示药物的潜在靶点,以及数种正在临床试验中的药物靶点,包括用于帕金森病的LRRK2抑制剂和用于心血管疾病的olpasiran。

慢性非传染性疾病是全球发病和死亡的主要原因,许多疾病由于缺乏有效的临床前模型和靶点证据的药物导致试验失败率高,因而缺乏有效治疗。自2005年首次全基因组关联研究以来,基于生物样本库的大规模基因研究揭示了与疾病相关的变异,结合ClinVar和OMIM等数据库的临床遗传学数据,为药物发现和精准医疗提供了宝贵的见解。2013至2022年FDA批准的新药中,有63%得到了遗传学证据支持,且有遗传学支持的药物机制成功率是无支持机制的2.6倍。作者最近的遗传优先评分(GPS)框架验证了将罕见和常见变异结合用于药物靶点优先排序的有效性。
然而,传统的基因关联研究依赖于二元病症标签,导致漏诊、误诊,无法根据风险和病情分层,统计能力有限。为此,近年来研究采用机器学习生成心血管、肺部和精神疾病的连续表型表示,从而识别出更多疾病相关变异,有望促进更有效药物的开发。本研究中,作者引入了GPS的机器学习辅助版本ML-GPS,应用于112种慢性疾病的表型代码。ML-GPS通过机器学习改进表型生成,使用连续表型表示基因与药物指示的关系,并结合13种遗传证据来源,给每个基因-表型对赋予药物指示概率,以便于进一步筛选高分基因。
ML-GPS有四大改进:1. 利用英国生物样本库的数据预测表型诊断,并对所有参与者进行再表型,以连续概率代表各表型;2. 使用连续编码反映靶点-疾病遗传关联的显著性或变异数量;3. 使用梯度提升模型代替逻辑回归,捕捉特征和药物指示之间的非线性关系;4. 采用phecodeX的子代码提高表型精度和疾病细分。
机器学习模型的构建用于预测表型诊断
作者筛选了phecodeX中的3612个表型代码,识别出336个与11类非传染性慢性疾病相关的代码。为识别与慢性生理变化相关的表型代码,使用LightGBM构建初步梯度提升模型,并且仅用年龄、性别和72项实验及生命体征数据。112个表型代码的模型表现达标,内分泌/代谢、血液/免疫和心血管类别表现最佳,而肌肉骨骼、皮肤和感觉器官类别表现较低。
对于表现合格的112个表型代码,作者构建了最终模型,增加了165项特征,包括生活方式、药物使用和诊断历史。LightGBM模型对特征选择具有鲁棒性,且优于或相当于XGBoost和随机森林模型。最终模型具有较高的区分能力和校准度。相比初步模型,最终模型在AUROC和AUPRC方面分别提高了0.08和0.06。此外,对于13个可通过单一生物标志物部分定义的表型代码(如肥胖——体重指数),最终模型在诊断表现上优于单一生物标志物。
112个表型代码中有70个的模型使用了三种或以上的特征类别。模型的重要特征一般与已知疾病特征一致,如铁缺乏性贫血的红细胞分布宽度和血红蛋白、痛风的尿酸和抗痛风药物使用。112个表型代码中有93个显著与全因死亡率相关,心脏骤停的风险最高(风险比为7.98),预测概率的分位数增高与全因死亡率显著相关,这表明预测概率与疾病风险、严重性、进展和漏诊相关。
遗传关联分析
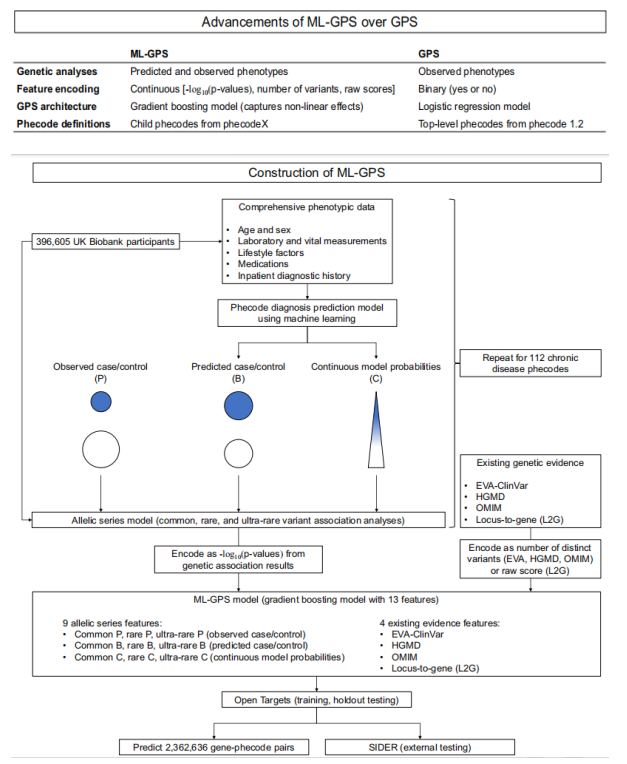
图 1
如图1所示,作者对每个基因-表型对建立了等位基因系列模型,进行了全基因组常见变异关联分析、外显子组单个罕见编码变异关联分析,以及基因水平的极罕见编码变异分析,分别针对观察到的表型状态(P)、二值化的模型概率(B)和连续模型概率(C)三种表型。
在罕见变异分析中,中位膨胀因子(λ)为1.04(P和B)和1.03(C);在常见变异分析中,中位λ为1.03(P)、1.06(B)和1.34(C);在极罕见变异分析中,中位λ为0.76(P)、0.89(B)和1.03(C)。C中常见变异的λ增大可能是由于多基因遗传下因果变异的增加。
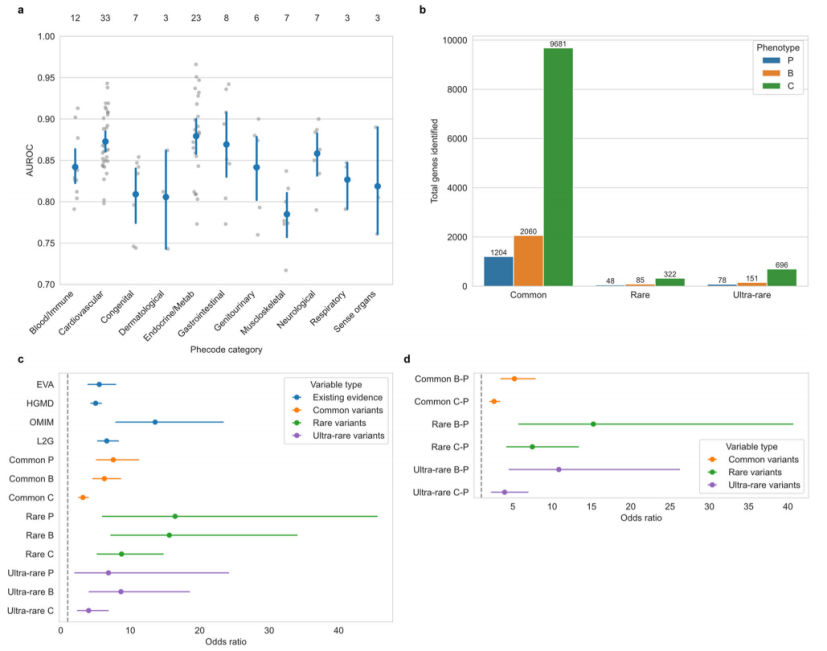
图 2
如图2b所示,在所有表型代码中,C识别的具有显著变异的基因数量远多于B,后者多于P。具体来说,常见变异分析中P、B和C分别识别了64、75和111个表型代码的基因;罕见变异分析中分别为40、46和108;极罕见变异分析中为53、61和109。B和C识别的基因与P的重叠率显著高于模型的AUROC,表明高区分度模型能更好代表观察到的表型。此外,在三种变异分析中,C识别的基因效果量中位数在罕见和极罕见变异中高于常见变异。
遗传特征与药物指示的关联
通过预测表型的遗传分析,B和C识别出具有药物指示的基因数量多于P。在常见变异分析中,C比P多识别出25个表型代码的基因,其中16个代码在P中未识别到。在罕见和极罕见变异分析中也表现出类似趋势。
与之前的报告一致,具有EVA-ClinVar、HGMD、OMIM和L2G证据的基因-表型对与药物指示显著相关,图2d中分别在Open Targets中对应OR为6.61、4.87、13.20和6.68。对于常见变异分析,P、B和C的OR分别为7.56、6.28和3.19。对于罕见变异,P、B和C的OR为16.46、15.62和8.75;对于极罕见变异,OR为6.87、8.66和4.02。即使在减去P识别的基因后,B和C仍然增加了药物指示基因的覆盖率。
ML-GPS构建
作者构建了机器学习模型以预测每个基因-表型对是否具有药物指示。在Open Targets和SIDER数据库中,分别包含112,274个和58,674个基因-表型对,其中分别有4116和1883个具有药物指示。模型纳入了13个特征,包括3个临床证据特征(EVA-ClinVar、HGMD、OMIM)、1个L2G特征以及9个来自P、B和C变异分析的特征。
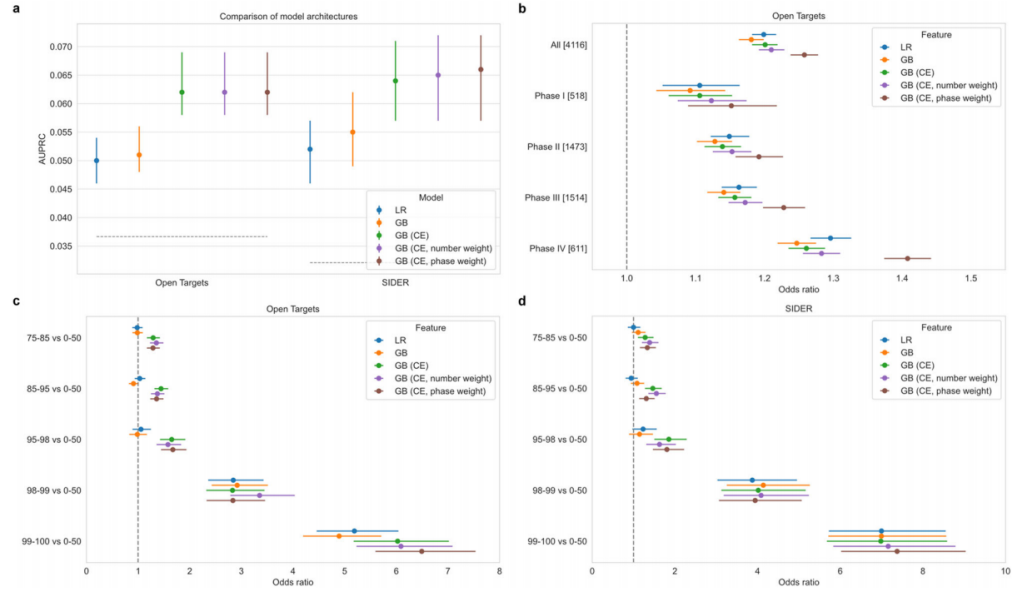
图 3
作者测试了五种模型结构,包括ElasticNet逻辑回归(LR)、梯度提升(GB)及其不同编码和权重版本。图3a显示,GB(CE, phase weights)模型在Open Targets和SIDER中的AUPRC表现最佳,优于LR和其他GB模型,其中L2G+Clinical+PBC特征组合在两数据集中提升显著。对L2G + Clinical + PBC模型的Shapley分析显示,B和C中的罕见及极罕见变异是重要特征,而OMIM特征因冗余而贡献较少。
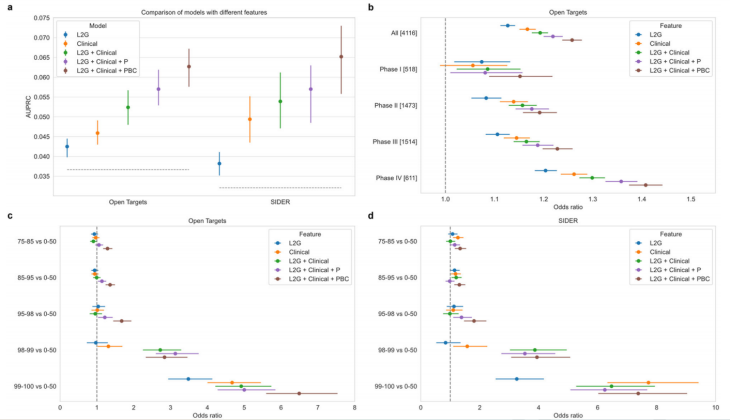
图 4
基于这些结果,作者采用L2G + Clinical + PBC特征组合下的GB(CE, phase weights)模型作为ML-GPS,并提供不同阈值下的精确度和召回率。尽管无法直接与原GPS模型对比,作者与包含L2G+Clinical+P特征的逻辑回归模型进行比较,图4中ML-GPS在AUPRC和OR方面均有提升,显示了更好的药物指示覆盖率。
带有作用趋势的ML-GPS 构建
作者扩展了ML-GPS,增加了作用趋势(Direction of Effect, DOE)预测。ML-GPS DOE是一个一对多分类器,为每个基因-表型对分配无药物指示、激活剂指示和抑制剂指示的概率,总和为1。与之前的GPS DOE不同,它不再仅输出正负分数。
在Open Targets和SIDER中,抑制剂指示多于激活剂指示。尽管训练时对激活剂指示赋予双倍权重,预测结果中抑制剂指示的AUPRC和OR仍较高。L2G + Clinical + PBC模型在激活剂和抑制剂指示预测中表现最佳。
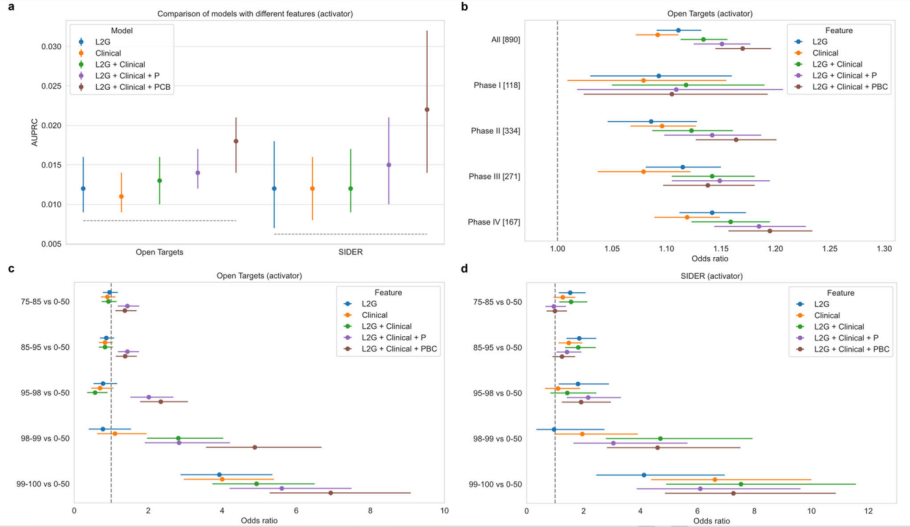
图 5
如图5所示,在激活剂指示预测中,L2G + Clinical + PBC模型在Open Targets和SIDER中的AUPRC分别为0.018和0.022,每标准差的分数增加对应OR分别为1.17和1.24。
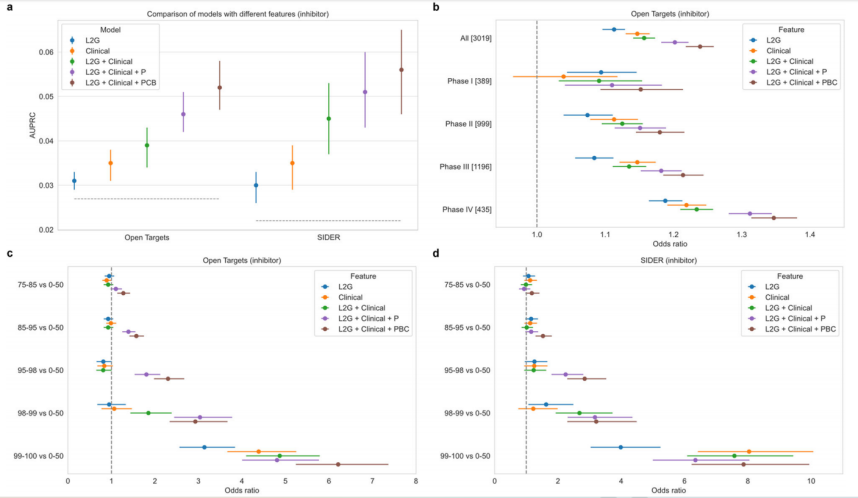
图 6
如图6所示,在预测抑制剂药物指示时,L2G + Clinical + PBC模型在Open Targets中的AUPRC为0.052,在SIDER中为0.056。在Open Targets中,每增加一个标准差,抑制剂药物指示的OR为1.24,而在99-100百分位与0-50百分位相比,基因-表型对在Open Targets中的OR为6.21,在SIDER中为7.87。基于这些结果,作者同样使用L2G + Clinical + PBC模型的得分作为ML-GPS DOE。
ML-GPS DOE提供了不同阈值下的精确度和召回率。例如,在激活剂指示中,阈值0.084的精确度和召回率分别为0.060和0.044;在抑制剂指示中,阈值0.204的精确度和召回率分别为0.250和0.022。
ML-GPS靶点和通路优先分析
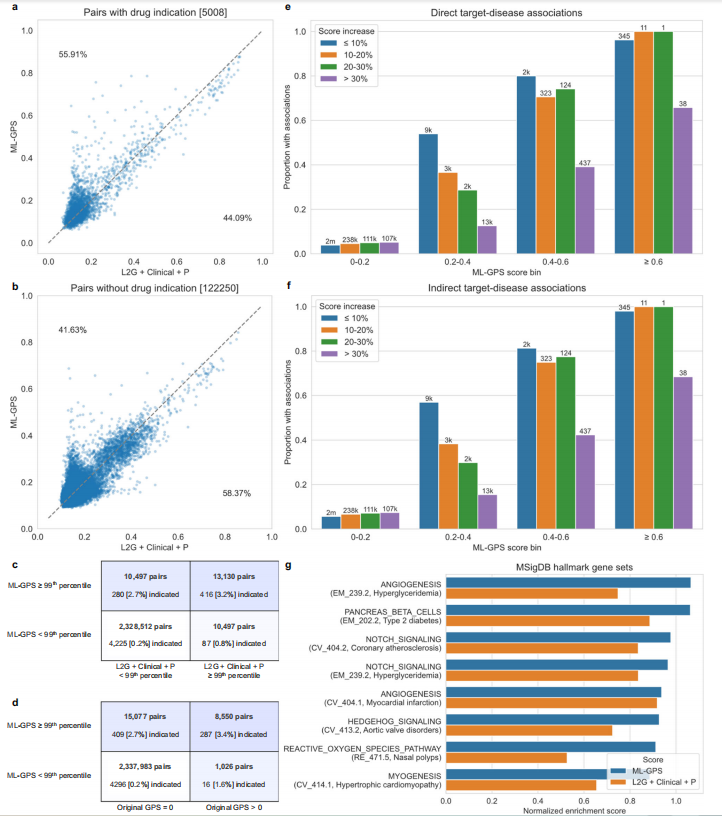
图 7
作者生成了ML-GPS和ML-GPS DOE对2,362,636个基因-表型对的预测,其中26,035个基因代表了不同的表型。如图7所示,在Open Targets和SIDER数据集中,ML-GPS比L2G + Clinical + P模型在标注药物指示方面表现更好,99分位数及以上的ML-GPS得分对应更多药物指示,表明使用C和B特征可改善药物靶点识别。
与原始GPS模型相比,ML-GPS覆盖了更大范围的药物靶点。ML-GPS识别的前23,626个基因-表型对中,26.1%有膜或分泌产物,50.6%具备理想的组织特异性,显示其在药物开发中的潜力。在ML-GPS得分提高超过30%的基因-表型对中,许多仅凭借C和B特征支持,例如GBA(帕金森综合征)、LDLR(高甘油三酯血症)和TMPRSS6(贫血),展示了这些靶点在疾病研究中的潜力。
此外,ML-GPS还支持多个正在临床试验的靶点,如LRRK2(帕金森病)、LPA(心血管疾病)和CFB(老年性黄斑变性)。通过单样本基因集富集分析,作者发现ML-GPS识别了与已知疾病机制一致的通路,并且揭示了原始模型未发现的疾病相关通路,如“未折叠蛋白反应”(先天性心脏病)和“血红素代谢”(铁代谢紊乱),进一步支持了ML-GPS的生物学和临床应用潜力。
讨论
本研究引入了ML-GPS,一种机器学习辅助的遗传优先评分框架,通过四大改进增强了112种慢性疾病的药物靶点识别。首先,ML-GPS结合遗传关联与机器学习预测的疾病表型,缓解慢性疾病漏诊问题。其次,采用连续编码方式将所有遗传关联纳入模型,优化特征的显著性阈值。第三,使用梯度提升方法捕捉特征与药物指示间的非线性关系。第四,采用改进的phecode术语表征疾病表型。ML-GPS显示出显著的药物靶点识别提升,并且支持多种临床试验中的创新药物。未来,ML-GPS有望扩展至更多群体和疾病领域,为精准医学和药物开发提供支持。
编译 | 于洲
审稿 | 王梓旭
参考资料
Chen R, Duffy Á, Petrazzini B O, et al. Expanding drug targets for 112 chronic diseases using a machine learning-assisted genetic priority score[J]. Nature Communications, 2024, 15(1): 8891.
内容中包含的图片若涉及版权问题,请及时与我们联系删除

评论
沙发等你来抢