
PK-Sim学习笔记
勃林格殷格翰研究团队在12月发表一篇有关人体PK预测的综述。不同于以往的推荐采用单一的方法预测PK参数,其推荐整合体内体外数据一起来预测PK参数和平均药时曲线。并基于公司内部的40个小分子进行方法验证,总体PK参数预测在2倍左右,平均药时曲线预测在3倍范围内。
本文对人体PK预测进行了全面的阐述,通过将体外的吸收(A)、分布(D)、代谢(M)、排泄(E) 和体内的PK数据以及专家判断相结合。提供了对PK预测结构化过程的详细评估和指导,预测超出单个参数的完整药时曲线。并讨论了未来的挑战和改进的方向,旨在为从事DMPK或者PKPD的科学家提供有价值的见解。
背景
药动学的探查一直是药物研发过程中的重点,良好的药动学行为是药物能成功上市的关键。药物研发过程中PK的预测也是一个逐步的过程,在获得第一个体外检测的数据或动物PK数据后,研发人员通常便可比较各个候选化合物,并决定未来的开发方向。二十年前,勃林格的药物研发人员发现仅关注CYP介导的代谢会导致人体PK预测错误判断(因为未考虑II相代谢酶),于是迭代建立人肝细胞孵育来计算肝脏清除率。但肝细胞孵育手段预测人体PK参数也存在不少缺陷,故其团队一直在探求更准确预测人体PK参数的方法。
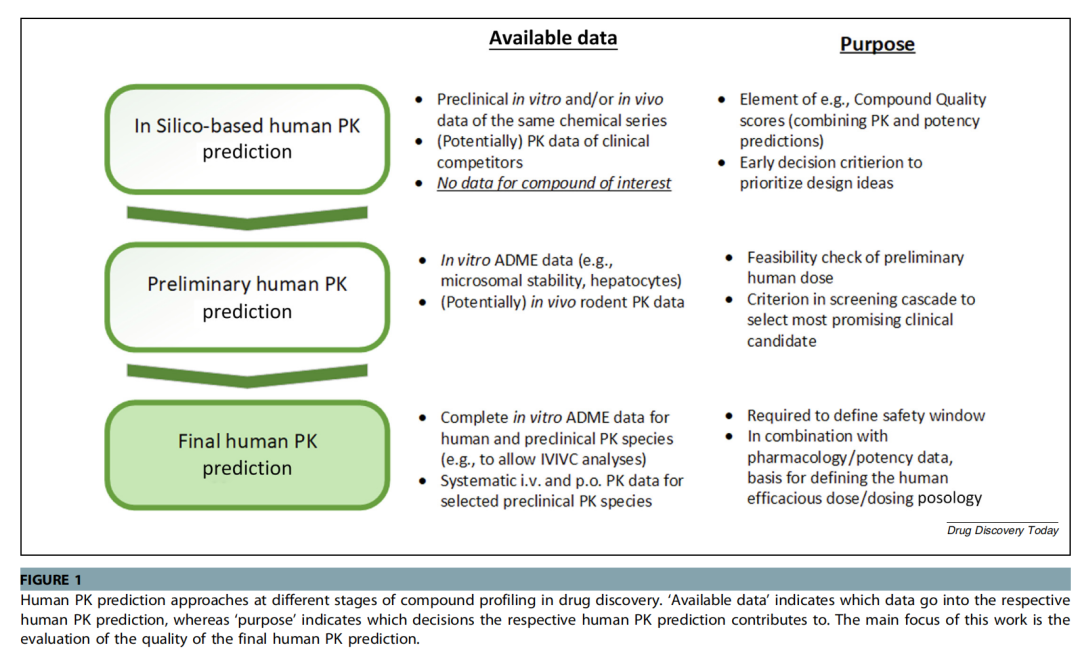
尽管肝脏是大多数药物的主要清除方式,但非肝清除也在药物的体内清除中占据不小的比例。所以预测表观清除率的方法非常重要,目前对人体PK预测进行了广泛的研究,但较多的文章多是关注于Cmax/AUC,而不是完整的评估血浆浓度-时间曲线,所以本研究对人体PK预测提供了一个详尽的指导,首先对关键PK参数的预测,其次是对药时曲线图的预测。
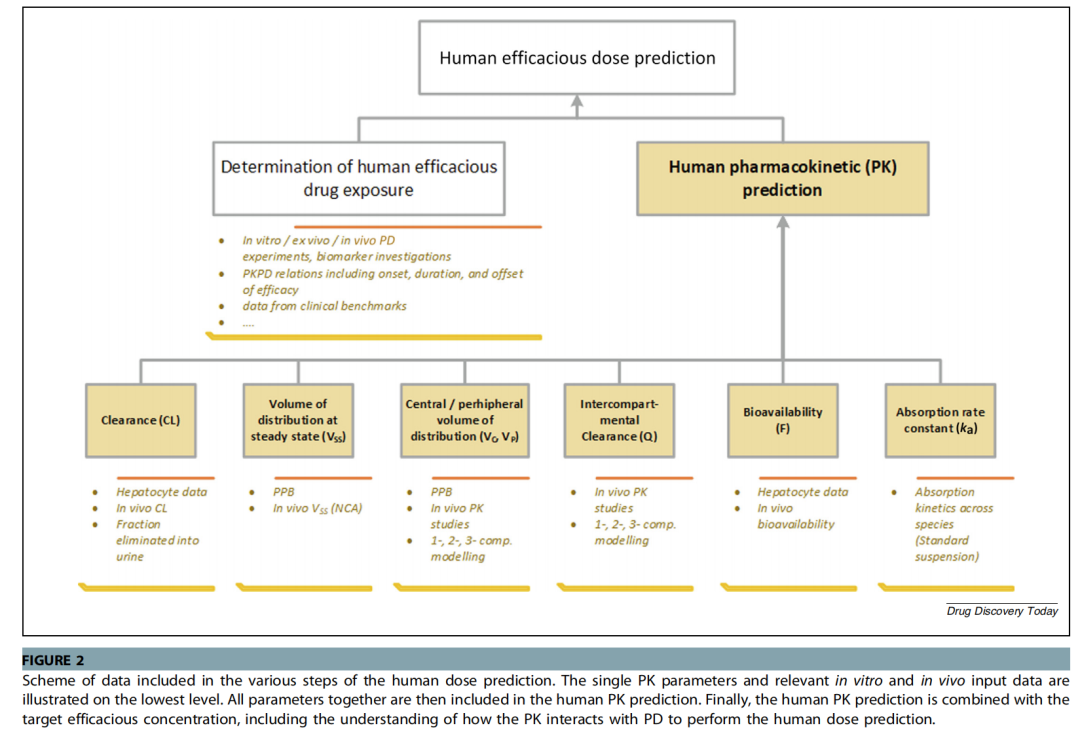
方法
数据集构建
临床血药浓度数据来自于40个口服化合物在健康志愿者的I期单剂量递增的临床试验中的数据。这40个化合物中25个符合类药5原则,15个化合物符合5原则中的4个原则。这些化合物主要是BCS1类和2类,有一种属于3b类,所有化合物均为固体制剂,没有静脉PK数据,故所评估的均为表观参数。
为了更好的预测人体PK曲线,数据集整合了所有化合物的临床前体外数据和体内数据,包括肝细胞稳定性和血浆蛋白结合数据,以及至少三个不同种属的体内PK数据。
关键PK参数预测
CL/F和Vss/F是药物中两个关键PK参数,对于这两个参数的预测,采用平均折叠误差和中值折叠误差来评估参数预测的质量。针对每个化合物单独选择最合适的方法,对于CL/F的评估,基于AUCInf 评估预测和实测的CL/F。对于Vss/F的评估,用MRT,CL/F以及Ka的模型拟合值计算临床的Vss/F,并与预测的进行比较。
清除率预测:
清除率一般=剂量/AUC,且与Vss/F一起,还可以提供静脉给药后的MRT的值。对于肝脏清除的药物,基于肝细胞的预测方法是首选方法,但实验室之间的差异可能会影响肝细胞清除率的测定,所以应使用跨物种的标准化测定装置来预测清除率。跨物种的CL通常与肾小球滤过率(GFR)或肾血流量(KBF)成正比,且这两个参数通常与跨物种的异速生长关系较好。
最佳的清除率预测方法由DMPK的专家决定,专家通过评估IVIVC的质量、跨物种代谢途径的潜在差异以及数据集的充分性和一致性等因素,来确保预测的质量。清除率的预测手段通常如下图所示:
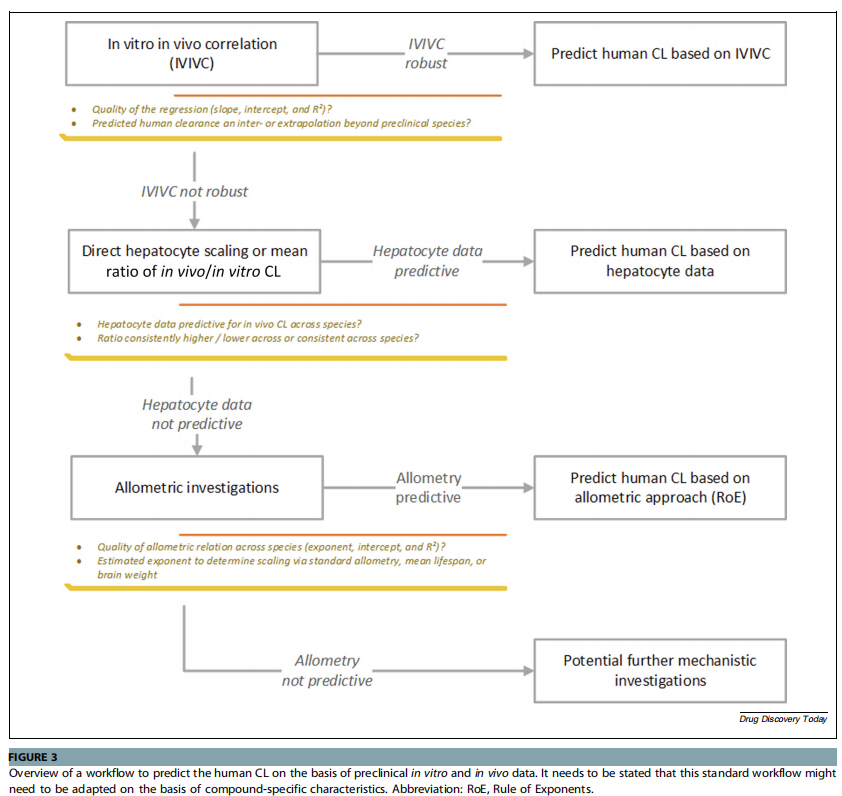
第一种IVIVC的方法至少需要三个物种的数据,这种相关性的建立是基于含有5%或者50%血清肝细胞孵育得出的肝提取比值以及静脉给药后计算出的相应体内清除率。根据血浆蛋白结合率大小和IVIVC的质量,选择基于5%或50%的血清相关性。图中所述斜率最佳为1,截距最佳为0。
第二种方法是体内/体外清除率的平均值比校正。基于临床前物种肝细胞的体内清除率和体外清除率之间计算比率,这种方法在IVIVC方法不能由单个临床前物种外推得到时非常有用,第二种方法的计算公式如下:
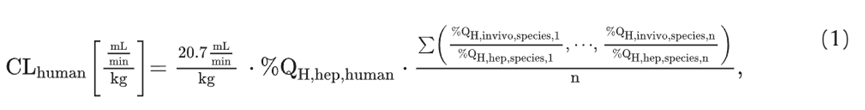
最后,当清除率与任何上述方法不一致且肝细胞不能预测体内清除率时,使用指数级的异速生长外推,对于主要经肾脏消除或者肝外代谢的药物,这种方法非常有用。
作者采用40个化合物进行验证,对12种化合物采用IVIVC进行预测,17种采用平均率比值预测,直接肝细胞缩放方法预测3种化合物,异速缩放方法预测8种化合物的清除率。也有一种化合物既有肾消除部分又有肝消除部分采用异速缩放和IVIVC联合外推的(理由是跨物种的主要消除途径不一致)。结果显示对CL/F,Vss/F,Cmax以及MRT的预测效果均较好。但结果也提示,没有任何一种独立的方法被认为能非常准确的预测所有化合物的CL(2倍范围内)。但直接从人类肝细胞进行缩放,结果显示预测性能不足,以及主要经肾排泄的药物,CL通常能较好的被预测。
分布容积的预测:
Vss(分布容积)是另一个非常重要的药代动力学参数。与清除率相比,它在不同物种间通常相对稳定。(前提是各物种之间对药物的血浆蛋白结合程度没有明显差异)药物在组织中的分布主要看它能多大程度地进入组织细胞中的脂质成分(比如中性脂类和中性磷脂),以及药物与组织中带负电荷的磷脂产生静电作用的能力(特别是带正电的药物分子)。即药物在组织里的分布与它的理化性质密切相关,比如药物的脂溶性、分子电荷状态等。
可以用Øie–Tozer等公式来估算分布稳态容积,这些公式通过已知的血浆中游离药物比例来预测。假设在组织中游离药物比例相对较大,而血浆中游离比例较小,那么药物更倾向保持在血浆中而不进入组织,使得Vss小于1 L/kg。反之,如果药物在组织中结合较多(比如深入细胞内,或在溶酶体等细胞内结构被“俘获”),那么Vss就往往会超过1 L/kg。简单来说,Vss反映了药物在体内血浆与组织之间的分布平衡。当药物更多分布在组织中(无论是游离药物还是结合形式),相对于血浆中的浓度,Vss 会增大。当药物主要集中在血浆中,而组织中分布较少时,Vss 会减小。
通常通过不同物种之间按体重标准化的Vss的平均值来进行预测。但是,结果显示用小鼠的数据来预测人类的Vss时,可能会低估真实值。因为小鼠的药动学非常快,可能会导致难以观察到终末期的药物浓度变化,从而造成对人类真实Vss的低估。所以,如果其他物种的Vss数据足够多,通常会把小鼠的数据排除在预测之外,以提高预测的准确性。简单来说,就是小鼠代谢太快,数据可能会“偏低”,所以我们更倾向于用其他动物的数据来预测人类的情况。
第二种预测方法是通过校正血浆中未结合药物的比例(fu)来预测Vss。如果在不同物种中, fu与Vss之间有明显的相关性,那么这种方法是首选。但如果按fu调整后的Vss没有明确的趋势,这种方法并不是最佳。
作为最后的核查步骤,通常比较非房室分析和药动学建模计算的Vss在不同物种中的差异。根据教科书的说法, NCA计算的Vss往往偏低,这是因为个体动物的数据不够完整,可能没有覆盖到足够长的时间范围。建模计算的Vss相对偏高,是因为在后期时间点,很多样本的浓度低于定量下限。例如,假设3只动物中只有1-2只的样本浓度高于定量下限,这会导致模型对药物分布的范围过度外推,从而高估Vss。简单来说,NCA容易低估,建模容易高估,关键在于数据覆盖的完整性和定量下限对结果的影响。
口服生物利用度的预测:
口服生物利用度(F)的计算受到多个因素的影响,包括药物从胃肠道吸收的比例以及逃过肠道首过效应和肝脏首过效应的比例。人口服生物利用度的标准方法为F = Fabs*Fhep。
外推的方法简要介绍为:将每个物种的估算的生物利用度除以Fhep(Fhep=1 – Eh,Eh:肝脏提取分数)来得到Fabs。然后将所有物种的Fabs平均,作为人类的Fabs值。最后根据人类肝细胞实验数据计算Eh,随后用1 − Eh得到Fhep即可得到生物利用度F。
预测药时曲线:
虽然CL/F和Vss/F在化合物筛选和候选药物选择过程中非常重要,但最终的人类剂量预测更多的是依赖目标暴露指标,比如某个特定的谷浓度、峰浓度或者暴露量。这些指标需要准确预测完整的药物浓度-时间曲线,而不仅仅是关键的PK参数。预测完整的PK曲线,除了CL/F和Vss/F以外,还需要额外的PK特征,如吸收和分布特性参数。因为即使Vss/F和CL/F相同,预测的浓度-时间曲线形状也可能有显著差异。
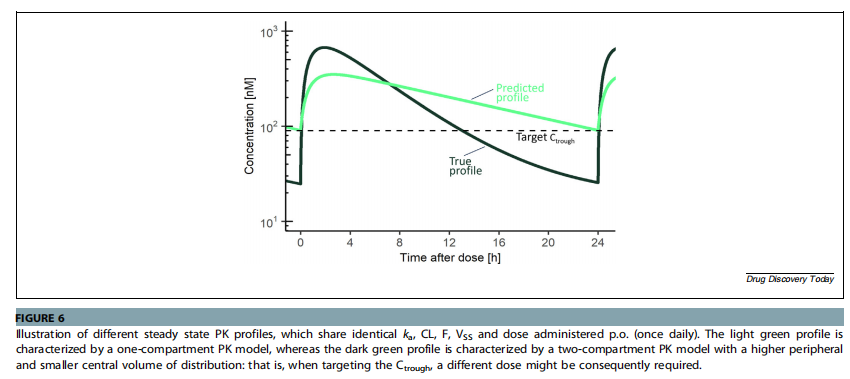
作者采用房室模型方法通过估算的PK参数(如中央和外周分布容积VC和VP、房室间清除率Q,以及吸收速率常数Ka)来考虑这些特性。此外,还有其他文献中提到的PBPK建模、Dedrick图以及Wajima叠加方法等可以用来预测浓度-时间曲线的形状。
房室模型参数的预测主要为以下方法:
1. 房室间清除率(Q):根据临床前数据估算,按固定的缩放比例(0.75)进行缩放,之后取缩放值的平均值用于人类PK预测。
2. 中央分布容积(VC):从各物种的建模数据中取均值,外周分布容积(VP)则通过公式VP = Vss(NCA) - VC计算。
3. 吸收速率常数(Ka):从各物种的PK建模结果中取Ka的平均值。但根据经验,小型猪(例如哥廷根小型猪)的吸收速率常数往往低估了人类的吸收速度,因此通常会将小型猪数据排除在Ka计算之外。
预测质量评估:
药时曲线预测评价指标:
1. 关键暴露指标的评估:重点评估C24h和Cmax等关键指标。
2. 曲线形状比较:为了比较预测和观察到的24小时内的血浆浓度-时间曲线,并避免因采样密度差异导致的偏差,使用了naïve pooled方法对观察数据建立PK模型,这个模型作为预测结果的直接对照。通过对预测和拟合浓度的几何平均误差倍数(GMFE)计算,评估预测质量,采样时间间隔设置为6分钟。
结果
评估结果:
Cmax:预测值表现出低估趋势,偏差为0.637。这可能与Vss预测误差相关。然而,65%的预测Cmax值在真实值的两倍范围内。
C24h:预测表现出更大的偏差,AAFE为33.4倍,MFE为2.45倍。这表明预测终末浓度(C24h)的难度更大。这符合预期,因为C24h受更多药代动力学参数的影响,而Cmax和AUC的影响因素较少。
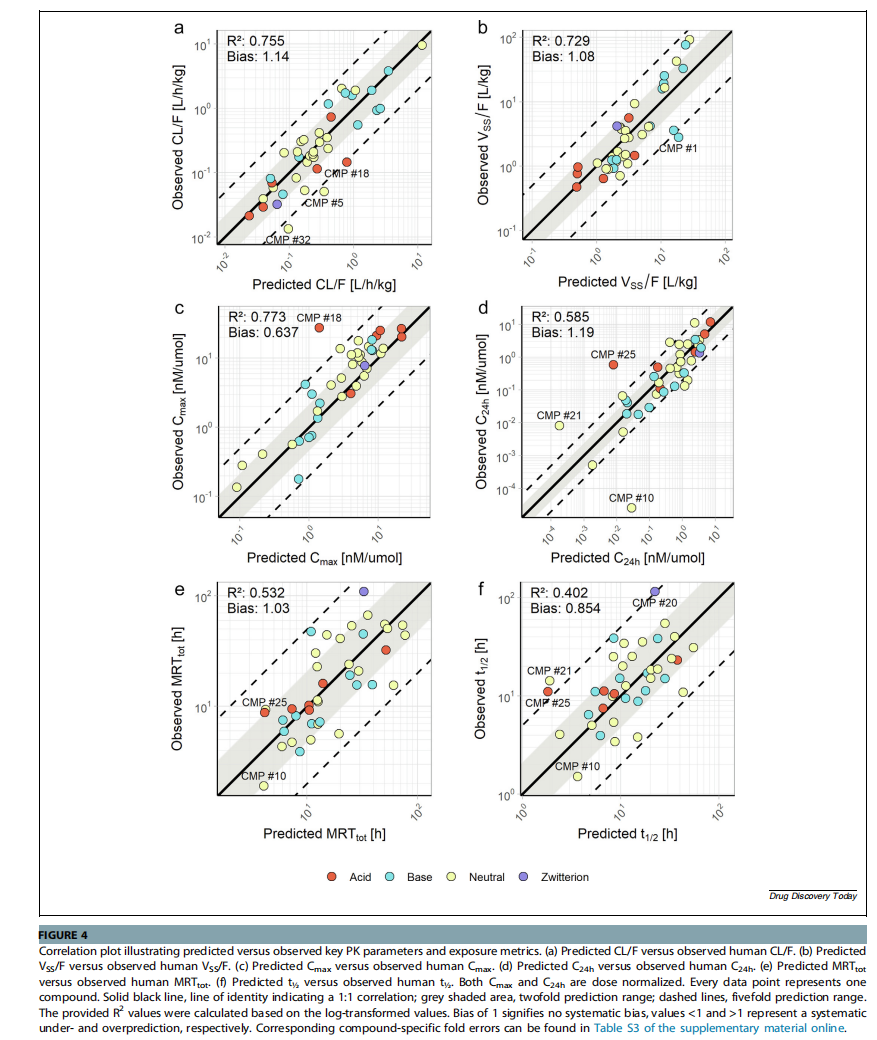
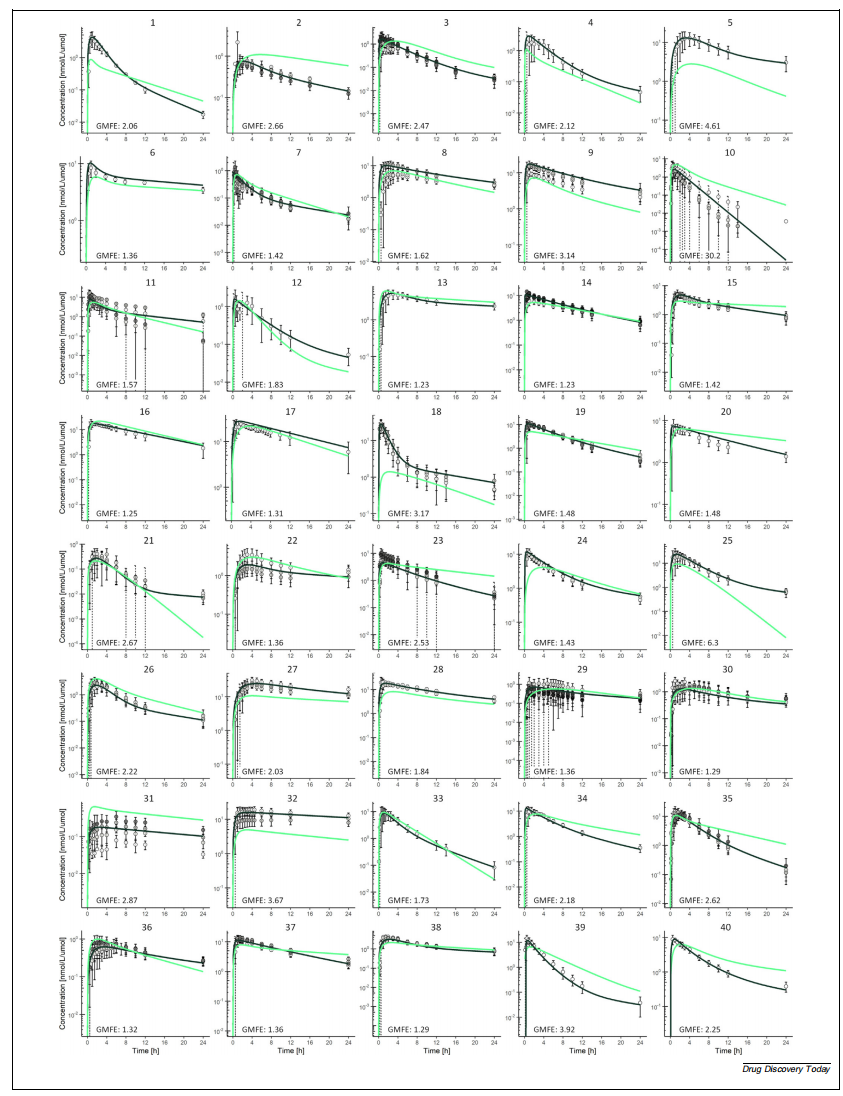
局限:
本文指出,当前的PK预测流程已能够为大多数化合物提供预测误差在两倍范围内的结果。然而,对于某些复杂情况(如超出Lipinski规则的化合物),可能需要采用定制化的预测方法。
相比之下,药物在人体剂量预测中的难点已逐渐从PK预测转向对有效浓度、生物标志物调控及其相关临床效果的准确评估。而这些评估的关键在于深入理解化合物的主要清除机制,从而选择最适合的预测策略。
参考资料
Himstedt A, Rapp H, Stopfer P, et al. Beyond CL and VSS: A comprehensive approach to human pharmacokinetic predictions. Drug Discov Today. Published online November 8, 2024. doi:10.1016/j.drudis.2024.104238
扫码入群


PK Sim学习笔记
内容中包含的图片若涉及版权问题,请及时与我们联系删除

评论
沙发等你来抢