
DRUGAI
今天为大家介绍的是来自宾夕法尼亚州立大学的Howard M. Salis团队的一篇论文。mRNA降解是影响基因表达水平的重要过程,但由于降解速率由多种相互关联的因素共同决定,仅通过序列预测mRNA的稳定性仍充满挑战。本研究对超过50,000条细菌mRNA进行了大规模的并行动力学降解测量,并采用“设计驱动学习”的方法开发和验证了一个预测mRNA稳定性的序列到功能模型。研究中设计的mRNA系统性地调整了其翻译速率、二级结构、序列组成,以及包含特定RNA结构的G-四链体和i-模体,同时调控RppH酶的活性,其半衰期从约20秒到20分钟不等。研究整合了生物物理模型与机器学习方法,构建了高精度、高通用性的mRNA稳定性稳态模型和动力学降解模型,并通过转录速率模型识别mRNA同工型,利用翻译速率模型计算核糖体对mRNA的保护效果。总体而言,该模型量化了细菌操纵子中共同影响mRNA稳定性的关键相互作用,并能够预测序列变化如何改变mRNA的稳定性。这对细菌遗传系统的研究与工程设计具有重要意义。

设计高性能遗传系统的关键在于建立更全面的预测平台,用于分析基因序列如何控制基因表达水平。然而,目前对于细菌mRNA降解速率的研究较为有限,而这正是影响蛋白质稳态水平和基因响应时间的关键因素。即使是很小的mRNA序列变化,也可能导致降解速率的10倍变化。因此,开发一个能够准确预测mRNA降解速率的模型,对代谢通路、基因电路、生物传感器和基因组的工程设计有重要意义。
mRNA的降解过程涉及多个相互关联的因素,如核糖体保护作用、mRNA结构(G-四链体等)、RNase结合位点以及RNA修饰酶的参与。作者通过设计62,120种合成mRNA序列,全面探讨这些因素的组合对降解速率的影响。作者在细菌中使用rifampicin处理后测量mRNA的降解速率,并结合生物物理建模和机器学习,构建了一个序列到功能的预测模型。这个模型不仅能准确预测mRNA的稳态水平和半衰期,还能识别5′ UTR中对降解影响最大的序列特性。
该模型基于过去对mRNA降解酶的研究,包括RNase E、RNase G和RppH等的作用机制。例如,RppH的去磷酸化活性对转录起始位点的前几个核苷酸非常敏感。此外,研究表明,未结构化的5′ UTR会加速mRNA降解,因为它为RNase E/G提供了更多结合位点。相比以往使用天然或小规模合成mRNA的研究,本研究通过大规模系统设计,解决了现有数据不足的问题,为遗传系统的精准工程提供了重要工具和方法。
5′ UTR序列的构建
研究人员大规模设计并构建了62,120个5′ UTR(信使RNA的5′非翻译区)序列,以研究其如何影响mRNA的降解速率。设计重点包括多个关键因素:(1) RppH结合位点,这是 mRNA 转录本的前四个核苷酸;(2)单链区域的长度和序列组成;(3)mRNA次级结构(如发夹、内部环等)的数量和折叠能量;(4)特殊的高级结构,如G-四链体和i-结构;(5)核糖体结合位点的翻译效率。
作者将这些序列分为多个实验组,每组分别测试不同因素的组合。例如,一组测试了1,280种5′ UTR,通过改变RppH结合位点和核糖体结合位点来覆盖10,000倍的翻译速率变化。其他实验组则重点探索单链RNA序列(如polyA, polyU)和特定结构(如发夹和G-四链体)对RNase结合位点的可及性和保护作用。
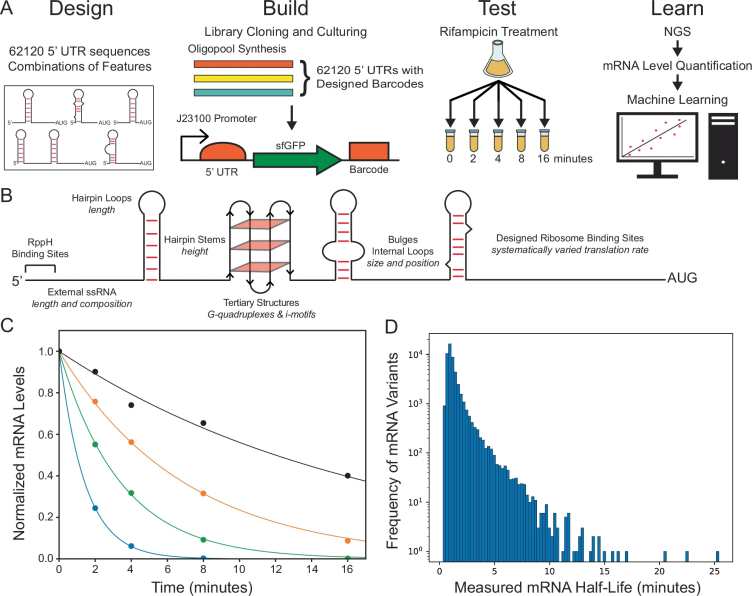
图 1
所有设计的5′ UTR通过170核苷酸的合成寡核苷酸编码,并附带独特条形码。作者通过两步克隆技术将它们整合到表达系统中,与报告基因sfGFP连接。虽然sfGFP本身并未用于测量蛋白质水平,但其核糖体结合速率为研究提供了一个可控环境。在E. coli中构建的质粒库通过MiSeq测序验证,结果显示覆盖率高达96.1%。图1A展示了核糖体结合如何影响mRNA的降解过程。
大规模平行动力学实验
为了研究设计的mRNA如何影响降解速率,科学家们进行了大规模平行实验,量化了62,120种mRNA的稳态和动力学降解速率。作者将混合细胞库在选择性LB培养基中培养,使细胞保持在指数生长期。作者随后加入rifampicin——一种抑制细菌转录的化合物——它停止新的mRNA合成,但保留了RNA降解机制。作者在多个时间点(T0, T2, T4, T8, T16)取样,通过RNA保护剂RNAprotect处理后提取总RNA。
为了确保不同时间点的数据可比性,作者在每份样本中加入已知浓度的对照RNA(spike-in RNA),并进行rRNA去除、cDNA合成和条形码扩增,最终通过Illumina NovaSeq测序获得超过16亿条读数。实验结果显示,91.5%的DNA条形码和93.5%的mRNA条形码在T0时的读数超过100,表明实验覆盖率非常高。随着时间推移,mRNA条形码的读数逐渐减少,符合指数衰减趋势。
作者使用公式M(t) = M0 e–kt(M0为初始mRNA水平,k为降解速率常数)对每种mRNA的降解进行拟合,并计算出半衰期(t1/2 = log(2)/k)。如图1C所示,不同mRNA的降解曲线差异显著,覆盖从0.31分钟到25.4分钟的半衰期范围,具体展现了生理相关时间尺度的多样性。图1D进一步展示了设计的5′ UTR序列如何显著改变mRNA的体内降解速率,为优化基因表达提供了宝贵的实验数据和模型验证。
多种因素决定mRNA降解
为了探究控制mRNA降解速率的多种因素,作者对设计的mRNA序列进行了深入分析。初步结果显示,单一因素(如RppH结合位点、翻译速率、5′ UTR的ssRNA数量)的影响较为有限,且同类别内部的差异可能超过类别之间的差异。因此,作者采用了多因素分析的方法,系统评估这些因素在不同条件下的综合作用。
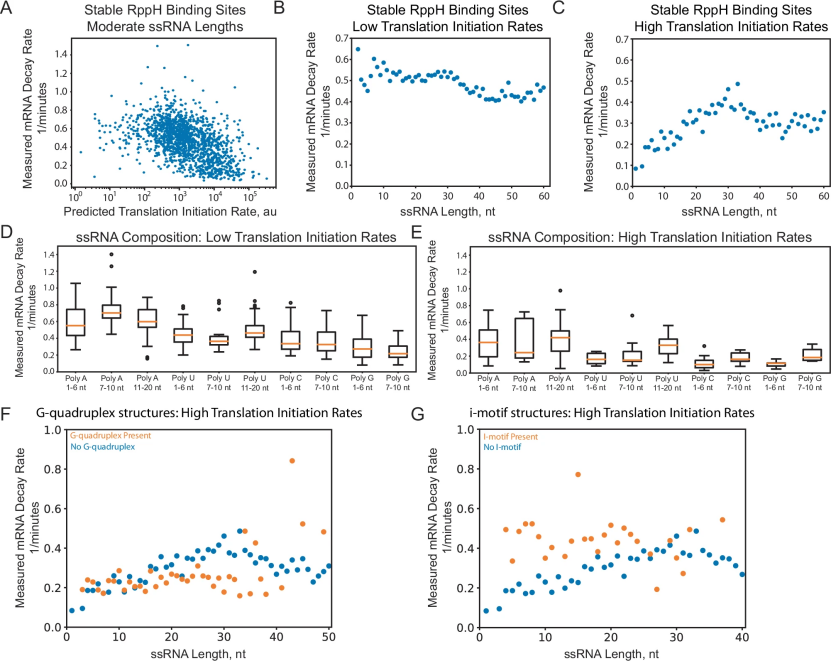
图 2
在多因素分析中,研究发现当RppH结合位点稳定且5′ UTR的ssRNA长度适中(11-40个核苷酸)时,翻译起始速率对mRNA降解速率的影响最为明显。数据显示,翻译速率较高的mRNA通常更稳定(图2A)。此外,ssRNA数量的增加显著提高了mRNA的降解速率,但这一趋势仅在翻译速率较高且RppH结合位点稳定的情况下清晰可见(图2B, C)。当翻译速率较低时,mRNA的降解速率本就较高,增加ssRNA对其影响较小。
序列组成对mRNA稳定性也起到了关键作用。例如,polyA序列的影响显著高于polyU或polyC,尤其在高翻译速率条件下,polyA导致更高的降解速率变化范围和分布(图2D, E)。此外,5′ UTR中的G-四链体结构展现了保护mRNA的作用,使其降解速率保持较低且稳定(约0.2 1/min, 图2F),而引入i-结构则显著加速了mRNA的降解(约0.5 1/min, 图2G)。
利用生物物理学和机器学习开发预测模型

图 3
作者开发了一种结合生物物理计算和机器学习的mRNA降解率预测模型,旨在揭示影响 mRNA 稳定性的关键设计因素(图3)。首先,作者列出了影响mRNA水平和降解率的潜在因素,包括启动子转录起始速率、RppH 结合位点、mRNA的结构类型和位置、5′ UTR 中单链RNA(ssRNA)的数量,以及CDS的翻译起始速率等。利用多个热力学模型(如Promoter Calculator v1.0和RBS Calculator v2.1),计算了这些特征的具体数值。此外,作者还采用RNA二级结构计算工具(如Vienna RNA v2.4.11)分析了mRNA结构特性。
通过实验发现,mRNA转录起始区(初始转录区, ITR)前四个核苷酸显著影响其降解速率,基于此,作者利用Promoter Calculator预测了最常见的mRNA异构体,并分别计算了它们的结构和功能特性。随后,作者采用LightGBM算法训练并测试模型,结果表明,该模型在预测mRNA稳态水平方面表现最佳,而在预测 mRNA 降解速率方面表现较为一般。关键特征如RppH结合位点、启动子转录速率和CDS翻译速率被证明对模型性能至关重要,而mRNA内部环长度等特征的重要性较低,模型也未因此获得显著改进。
控制mRNA衰减率的设计规则
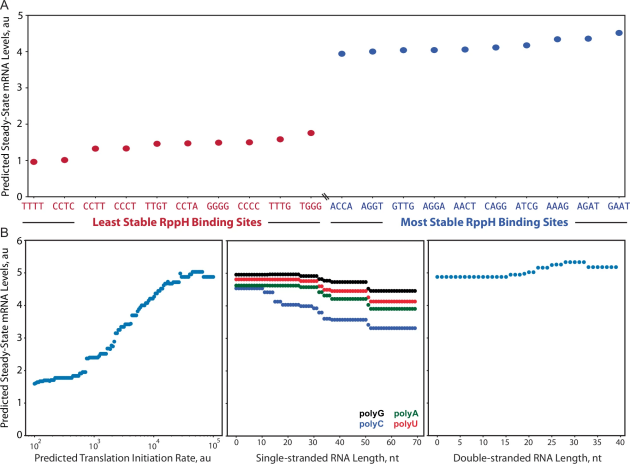
图 4
作者通过已开发的LightGBM模型,对mRNA的降解速率设计规则进行了深入分析,明确了多种独立因素对 mRNA 稳定性的作用机制(图4)。作者选取了一个具有高稳定性的基准mRNA,通过系统性调整其特定特征值,分析了这些变化对mRNA稳定性的影响。
研究结果显示:
RppH结合位点:结合位点的碱基组成对mRNA稳定性有显著影响,尤其是由相同核苷酸组成的结合位点(即同质性结合位点)会显著降低稳定性(图4A)。
翻译起始速率:翻译起始速率与mRNA的稳态表达水平呈S型关系。提高速率会增加蛋白质表达量,但在特定点后达到平台,不再有显著提升(图4B左)。
5′ UTR单链RNA:5′ UTR中较长的单链RNA会降低mRNA稳定性,这种影响在富含C或A的序列中尤为明显。相比之下,聚腺苷酸链因其刚性更容易被RNase降解(图4B中)。
mRNA二级结构:增加mRNA的二级结构对其稳定性影响较小,真正关键的因素是未折叠RNA的存在或缺失,而非二级结构的数量(图4B右)。
讨论
本文研究设计了62,120种5′ UTR序列,利用条形码细胞文库和深度测序定量测量mRNA稳态水平和降解速率,发现UTR序列特性显著影响mRNA半衰期(20秒至20分钟)。通过整合生物物理模型和机器学习(LightGBM算法),构建了预测mRNA稳定性的模型,解析了关键序列-功能关系。研究揭示了四类主要影响mRNA降解的机制,包括5′端序列调控RppH结合、翻译启动速率对mRNA稳定性的非线性影响、非结构化单链RNA对RNase易感性,以及G-四链体结构增强mRNA稳定性。这些机制在细菌间具有保守性,为基因表达调控提供了新见解,并开发了可用于设计遗传系统的mRNA稳定性计算器和在线平台。
编译|于洲
审稿|王梓旭
参考资料
Cetnar D P, Hossain A, Vezeau G E, et al. Predicting synthetic mRNA stability using massively parallel kinetic measurements, biophysical modeling, and machine learning[J]. Nature Communications, 2024, 15(1): 9601.
内容中包含的图片若涉及版权问题,请及时与我们联系删除

评论
沙发等你来抢