
DRUGAI
人类基因组中大部分被转录为RNA,这些RNA中包含许多对其功能至关重要的结构元素。这类RNA分子,包括那些具有明确结构和良好折叠的分子,通常具有构象异质性和灵活性,这是其功能发挥的前提条件。然而,这种特性限制了核磁共振(NMR)、晶体学和冷冻电子显微镜等方法在结构解析中的应用。此外,由于缺乏大型RNA结构数据库,以及序列与结构之间没有明确的相关性,类似AlphaFold用于蛋白质结构预测的方法并不适用于RNA。因此,解析异质性RNA的结构仍是一项未解决的挑战。在此,研究人员报道了一种结合原子力显微镜、无监督机器学习和深度神经网络的整体RNA结构解析方法(HORNET)。该方法利用溶液中单个分子的原子力显微镜图像,解析RNA的三维拓扑结构。由于原子力显微镜具有高信噪比,这种方法特别适合捕获处于不同构象的大型RNA分子的结构。通过六个基准案例,我们验证了HORNET的实用性,并成功解析了RNase P RNA和HIV-1 Rev响应元件(RRE)RNA的多种异质性结构。因此,该方法解决了大型柔性RNA分子异质性结构解析的主要挑战之一,并对RNA结构生物学的基础研究作出了贡献。

RNA结构和动态的研究对于理解其功能、设计新型RNA装置以及开发RNA靶向化合物具有重要意义。自近半个世纪前首次解析tRNA的三维结构以来,许多稳定的RNA结构已通过NMR、X射线晶体学和冷冻电子显微镜等方法得到解析。然而,这些方法依赖于信号在相对均一样本上的平均化,不适合研究在生理条件下具有高度异质性、功能动态性且不存在单一稳定构象的RNA分子。
在生理相关条件下的研究表明,RNA即使在保持结构的情况下,仍可呈现多种异质性构象。这种构象异质性与未折叠或本质无序的蛋白质不同,是功能性RNA的特征,也是其在细胞环境中与各种配体相互作用的前提。因此,单一静态的RNA结构无法准确描述其功能相关的构象变化。在RNA研究快速发展及其广泛应用于生物医学和公共卫生领域的背景下,迫切需要开发一种研究RNA高度异质性构象空间的方法。
原子力显微镜(AFM)的拓扑成像能够以高信噪比直接提供全局结构信息,在分辨双螺旋沟槽的同时保持无失真。此前研究已表明,全局结构信息对RNA结构预测的约束具有重要作用,且AFM可在生理溶液条件下可视化RNA的异质性构象。然而,目前尚未明确拓扑AFM图像与底层原子级拓扑结构的定量关系,也缺乏用于解析单个RNA构象并进行精度估算的完整方法和相关软件。
研究人员开发了HORNET,一种创新的RNA构象三维拓扑结构解析方法。该方法通过动态拟合,结合AFM伪势能和经典吉布斯自由能描述驱动模型的构象轨迹收敛。轨迹结构随后通过无监督和监督深度学习进行聚类和评估,综合考虑所有能量与拓扑信息。研究人员设计了一种深度神经网络(DNN)架构,基于伪结构数据库(psDatabase)进行训练,并通过六个基准测试案例验证其精度,提供顶级结构的均方根偏差(r.m.s.d.)评估。研究人员进一步应用HORNET,成功解析了全长RNase P RNA的三种新结构和HIV-1 RRE的五种新构象。
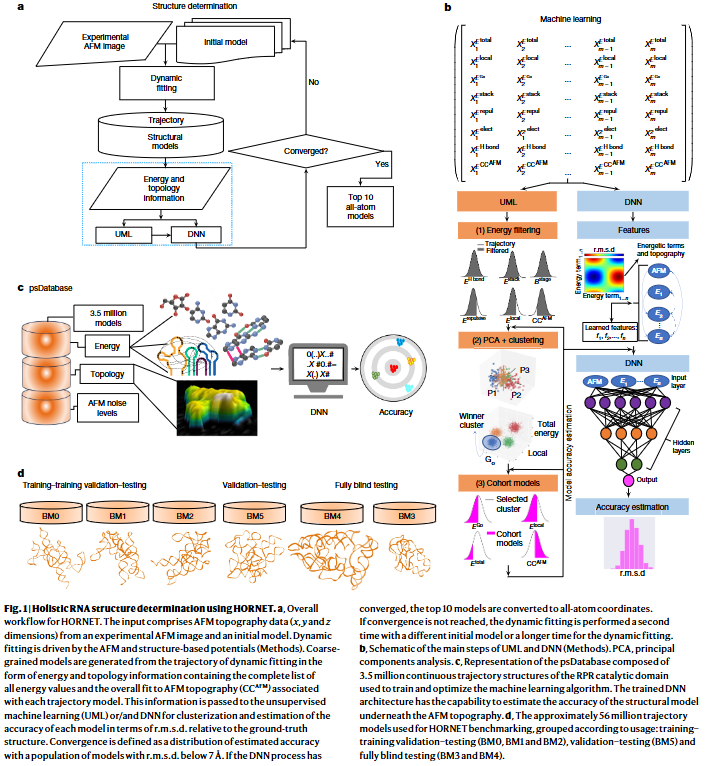
从AFM粒子图像到3D结构
以嗜热芽孢杆菌的RNase P RNA (RPR)为例,研究人员展示了通过AFM记录的单个RNA分子的图像。图像显示了三个不同构象状态下的RNA分子(P1、P2和P3),其构象均与晶体结构不同,交叉相关(CCAFM)得分分别为0.77、0.80和0.87。粒子拓扑背景噪声为最大z高度的1%至5%。通过应用低通傅里叶滤波器,P1、P2和P3图像的分辨率分别为0.87 nm⁻¹(11.5 Å)、0.90 nm⁻¹(11.1 Å)和0.80 nm⁻¹(12.5 Å)。
尽管约12 Å的分辨率看似限制了AFM在结构解析中的应用,但RNA结构的特点使AFM成为研究异质性RNA结构和动态的理想工具。首先,RNA折叠是分层和模块化的,使拓扑空间信息易于辨别;其次,大多数RNA结构由A型双螺旋组成,其主次沟槽尺寸在AFM分辨率范围内;第三,这些高度保守的A型双螺旋骨架占RNA结构质量的70%以上,其r.m.s.d.波动范围约为1.5 Å。因此,在初始结构模型中加入共价键连接和二级结构信息作为先验知识,可以从AFM分子表面重建3D拓扑结构,其不确定性显著低于AFM数据本身的分辨率限制,类似于低分辨率电子密度图的解析方法。
值得注意的是,目前尚无仅基于单个大分子信息解析出的生物大分子结构,已报道的结构均依赖信号平均方法。此外,RNA结构的稀缺性难以覆盖RNA可能采样的广泛构象空间。实际上,只有四类裸RNA(大于210 nt)在3.5 Å以下分辨率内有记录:腺苷钴胺核开关、I型和II型内含子以及RPR。RPR是一种多轮催化核酶,处理pre-tRNA和其他RNA的5′端,表现出已知的构象灵活性和多样结构特征。
为了建立方法,研究人员使用RPR的催化核心域(PDB ID: 3DHS)生成初始模拟数据(称为BM0),通过施加7个不同水平的高斯噪声(最大z高度的5%、10%、15%、20%、30%、40%和50%)动态拟合得到。动态拟合采用经典Langevin粗粒化分子动力学模拟,结合AFM伪势能进行广泛构象空间采样。AFM拓扑约束效应通过残基动态交叉相关图(DCCM)体现,其中AFM约束的DCCM仅是自由(无约束)DCCM的一个子集。随着噪声水平增加,相对于3DHS的最佳结构(最低r.m.s.d.)的数量减少,r.m.s.d.最低值从2.97 Å(5%噪声)增加至6.04 Å(50%噪声)。
从无监督机器学习(UML)中筛选的最佳结构
对BM0轨迹模型的r.m.s.d.值分析表明,大量模型接近真实结构(3DHS),但它们的能量与其他构象相似,即使这些结构与真实结构有显著差异。因此,当真实结构未知时,仅基于能量的传统统计方法不足以识别AFM图像中接近真实结构的模型。同样,分子表面与结构之间的交叉相关(CCAFM)得分也不足以单独识别最佳结构模型,因为高CCAFM得分可能以牺牲结构完整性和分层折叠原则(过拟合)为代价获得。
研究人员使用整体无监督机器学习(UML),将以下三种信息组合作为输入:(1)与初级化学结构、二级和三级结构相关的能量;(2)CCAFM得分;(3)与AFM拓扑约束相关的能量成本(AFM偏置势)。这些信息完全来源于结构模型和AFM拓扑图像,且没有任何假设成分。
首先,通过初始能量过滤去除轨迹中的离群值,然后进行两步UML分析:对所有能量项进行主成分分析(PCA),并通过连续聚类算法识别最低能量分布的模型集群。聚类过程中使用的Go势包含反映模型折叠程度的信息,但动态拟合中Go势的权重设置为最低值,以确保AFM偏置势能够采样显著不同于初始结构的构象。从最佳UML集群中,选择既具有最低能量(EGo、Elocal和Etotal),又具有最高CCAFM的模型群。
UML管道通过迭代方法筛选出r.m.s.d.最低的子模型群。在BM0的所有测试噪声水平下,几乎所有最佳模型都包含在最终UML集群中。即使在最高噪声水平下,前10个选择模型的平均r.m.s.d.仍约为5 Å,说明拓扑约束在全局构象分析中的优势。与任何结构拟合方法一样,动态拟合的效果高度依赖于初始模型。我们的基准测试(BM0–BM5)涵盖了不同RNA以及通过多种方法(结构预测、轨迹模型和小角X射线散射(SAXS)数据)获得的不同初始构象。结果显示,在所有基准测试中,UML集群中总能量最低的前10个模型平均r.m.s.d.约为5 Å,相对于真实结构的最低r.m.s.d.约为3.5 Å。
使用深度神经网络(DNN)估计模型准确性
尽管UML可以筛选出最佳模型群体,但无法估计每个模型相对于AFM图像下真实结构的准确性,而这种估计对解析未知结构至关重要。由于重建结构的准确性嵌入在模型(能量项)和AFM拓扑(CCAFM)中,经过良好训练的DNN可以在真实结构未知的情况下提供准确性估计。DNN用于估计准确性(置信水平)已在最新的蛋白质结构预测中得到验证,这依赖于数据库中丰富的结构信息和序列-三维结构的相关性。然而,由于RNA缺乏丰富的结构数据以及序列-结构相关性,RNA结构预测的难度更大,尤其是在分子较大且具有高度构象异质性时,因为不同构象之间的几何和能量等价性以及缺乏全局约束进一步增加了挑战。此外,同一RNA序列可能折叠为截然不同的构象,因此实验获得的单分子数据对解析单个构象的结构至关重要。
psDatabase的构建与DNN训练
为了应对实验确定的RNA结构不足的问题,研究人员创建了一个包含超过350万个RPR催化结构域模型的伪结构数据库(psDatabase),其中包括约150万个BM0轨迹模型,以及BM1和BM2分别约100万个模型。这些轨迹模型覆盖了连续的构象空间,优于离散片段数据库,且其模型间的r.m.s.d.差异可达37 Å,反映了构象空间的广度。我们将psDatabase的80%用于DNN训练(训练集),20%用于初步验证和测试(训练验证集),以控制正则化过程中的欠拟合和过拟合。为评估DNN是否对不同RNA轨迹及真实结构具有泛化能力,我们将BM5的5%用作进一步的验证数据集,这种验证方法被认为比包含所有基准数据集部分数据的方法更现实且稳健。
DNN的训练与验证
在训练验证集中,DNN的预测r.m.s.d.值与真实值的相关性极高,Pearson系数达到0.95。训练与验证集的损失函数随着迭代次数(epoch)的增加而下降,表明模型在50个epoch内逐步学习,并在300个epoch内未出现欠拟合或过拟合。在验证集中损失函数最小的epoch用于确定训练的停止点。
DNN的测试与泛化性能
为评估HORNET对不同轨迹和RNA分子(形状、大小和序列不同)的性能,研究人员对BM1–BM5的完整轨迹进行了测试。结果表明,DNN学习的是通用的结构准确性“度量”,而非特定结构特征的记忆。尤其是对BM3和BM4的盲测显示,DNN架构的结果与UML的结果一致,并交叉验证了最佳模型群体的准确性估计。这进一步证明,DNN不仅可以可靠地从未见数据中预测准确性,还可以为UML筛选出的顶级模型群体提供准确性的信心评估。
验证不同初始模型的准确性
建立DNN流程后,研究人员验证了HORNET是否能够基于相同的AFM拓扑图像解析结构,但采用两种不同的初始构象。对于BM1 (S142) 和BM2 (S1076),初始RPR催化结构域模型由FARFAR2(Rosetta的RNA片段组装与全原子精细化)生成,并具有ARES评分分别为9.23和9.04。这两种初始结构是从FARFAR2模型池中选择的,结合了最佳ARES和FARFAR2评分,同时满足相对于晶体结构(PDB: 2A64)的r.m.s.d.阈值(10 Å和20 Å)。BM1和BM2的初始r.m.s.d.分别为13.5 Å和22.3 Å,BM1与BM2之间的r.m.s.d.为18.7 Å,表明两者的拓扑结构彼此之间以及与真实结构均显著不同。在每种情况下,UML和DNN均成功识别出最佳模型群体,并展示了DNN预测与实际r.m.s.d.之间的高度相关性(BM1为0.92,BM2为0.80)。
对于未收敛的初始模型(如S257,ARES评分最佳但陷入局部最小值),通过无约束分子动力学模拟将其驱出局部最小值后,再应用UML动态拟合。结果显示,通过HORNET的最佳DNN模型群体能够收敛到接近真实结构的模型,最低r.m.s.d.为3.6 Å。
使用小RNA数据进行验证
对210 nt的钴胺核开关(rCbl, BM3)的测试展示了HORNET对完全不同序列、形状和折叠RNA的能力。BM3的初始模型来源于粗粒化分子动力学轨迹,r.m.s.d.为10.2 Å(相对于真实结构PDB: 4GMA)。通过HORNET,UML和DNN筛选的模型群体显示出平均约3 Å的r.m.s.d.,DNN模型在整个轨迹中表现出良好的相关性(Pearson系数为0.84)。
使用SAXS派生模型的验证
BM4和BM5使用低分辨率实验技术生成的初始模型进行测试。BM4(395 nt)和BM5(298 nt)的初始模型来源于SAXS模拟数据,分别具有16.1 Å和14.0 Å的初始r.m.s.d.。通过HORNET筛选,UML的最佳模型群体的最低r.m.s.d.分别为3.8 Å(BM4)和5.4 Å(BM5)。DNN对BM4和BM5的相关性分别为0.77和0.64。
异质性构象的结构解析
HORNET应用于全长RPR的AFM粒子图像(P1、P2和P3),显示出三种高度异质的构象,与晶体结构显著不同。动态拟合轨迹揭示了P1、P2和P3的构象差异:P1采样了广泛的原子位移范围,P3呈中等采样,P2受限位移可能与AFM图像提供更多短距离信息有关。在HORNET的UML和DNN流程后,P1、P2和P3的最佳模型的预测r.m.s.d.在4–6 Å范围内。值得注意的是,P1的预测r.m.s.d.分布范围较广(5.6–31 Å),而P2和P3的分布较窄(分别为4.4–27 Å和4.7–12 Å),说明模型能够区分并评分不同范围的原子位移。
总的来说,HORNET成功验证了其在不同RNA序列、形状和初始模型条件下的泛化能力,能够可靠地解析异质性RNA的三维结构并提供准确性估计。
HIV-1 RRE RNA的几种构象解析
许多功能性RNA具有高度动态特性,在生理条件下不采取单一稳定结构。HIV-1 RRE RNA就是其中之一。病毒未剪切和单剪切转录本的核输出是HIV-1病毒复制周期的关键步骤,而RRE RNA是病毒在宿主RNA中识别并选择自身RNA进行输出的核心。尽管基于SAXS数据的RRE分子包络已被间接推导,但尚未获得RRE任何构象状态的高分辨率结构。因此,RRE的结构及其与Rev蛋白结合模式一直是热议的话题。由于RRE的构象异质性,晶体学和冷冻电子显微镜的适用性受限,进一步引发了关于病毒如何在RRE和Rev蛋白二聚体均灵活的条件下特异性识别RRE的根本性问题。
研究人员利用HORNET解析了通过AFM观察到的五种RRE构象,展示了其解析未知RNA三维结构的能力,这些结构与用于基准测试的RNA显著不同。AFM可视化显示,RRE折叠成多种“A”形状,验证了此前基于SAXS数据推导的分子包络。此外,在所有五种构象中,两个已知的Rev结合位点彼此相对,间距在45至70 Å之间,进一步揭示了RRE的构象异质性和灵活性。最大的构象灵活性和异质性集中在III–V结构域的100至190号残基之间,该区域曾被认为通过采用替代构象抵抗RevM10突变体对病毒输出的破坏。
鉴于RRE的构象异质性,特别是在结合位点之间的距离变化,研究人员设计了一类新型分支肽模拟Rev二聚体。该分子由两个通过赖氨酸主链和侧链并联连接的精氨酸富集基序(ARMs)组成。赖氨酸的八个单键连接确保了最大灵活性,使并联ARMs能够采样广泛的双螺旋角度和结合位点间距。这种设计实现了与RRE两个结合位点的拓扑互补性,同时分支肽和RRE的构象灵活性允许相互适应,从而达到最佳匹配。
实验表明,这种分支肽在高浓度Rev或非特异性RNA存在的情况下,仍能以高特异性和高亲和力结合RRE。结合物在电泳迁移率实验中显示更快迁移,表明其结构更紧凑、更少灵活性,与AFM图像和HORNET结构动力学结果一致。这种“A”形状的直接可视化明确解决了围绕RRE拓扑结构的长期争议。
拓扑互补性和相互构象适应性可能解释了HIV-1病毒如何在RRE具有灵活和异质性构象的情况下实现特异性识别。分支肽的成功结合为理解RRE与Rev之间的相互作用提供了新的视角,同时也为抗病毒药物设计提供了潜在的策略。
讨论
HORNET通过摆脱对信号平均的依赖,解决了研究高度异质性和柔性RNA分子拓扑结构的主要挑战,这是核磁共振(NMR)、晶体学和冷冻电子显微镜(cryo-EM)等方法的常见局限。尽管HORNET作为现有高分辨率方法的补充工具,其从单个RNA构象的AFM图像中重现拓扑结构的能力显著扩展了我们对RNA三维构象空间的认识,超越了数据库中少量静态结构的片段。鉴于RNA中丰富的结构元素,HORNET有潜力加速对具有生物学意义的大型RNA构象空间的理解,这一点在对HIV-1 RRE的研究中得到了体现。
这项研究中揭示的拓扑互补性和相互构象适应性可能是驱动RRE–Rev相互作用的普遍机制,解释了其特异性和高亲和力的来源。此外,估计未知构象结构的准确性一直是结构生物学中的一项重大挑战。蛋白质结构预测的最新进展以及RNA结构预测领域的进步令人振奋。由于RNA的构象异质性,像HORNET这样结合单个构象特异性拓扑全局约束的方法,为研究柔性RNA的构象景观提供了一种可行的途径。
拥有足够覆盖广泛RNA构象空间的结构数据库,并结合实验拓扑信息和二级结构信息的条件下,经过充分训练的DNN模型可以使HORNET在溶液条件下生成单个大型RNA构象的低分辨率拓扑结构。这种方法不仅为解析复杂RNA的动态结构提供了强有力的工具,还为深入理解RNA功能与结构的关系打开了新的研究方向。
整理 | WJM
参考资料
Degenhardt, M.F.S., Degenhardt, H.F., Bhandari, Y.R. et al. Determining structures of RNA conformers using AFM and deep neural networks. Nature (2024).
https://doi.org/10.1038/s41586-024-07559-x
内容中包含的图片若涉及版权问题,请及时与我们联系删除

评论
沙发等你来抢