
纠错是三代测序数据分析的第一步。但现有的方法往往难以处理嘈杂的数据,或者是为 PacBio HiFi 等技术量身定制的。针对 Nanopore R10 simplex 读数进行优化的方法还存在空白,这种读数的错误率通常低于 2%。2024年12月19日,湖南大学生物学院罗宵教授和湖南大学信息科学与工程学院刘元盛副教授合作在Communications Biology 杂志在线发表题为“Repeat and haplotype aware error correction in nanopore sequencing reads with DeChat”的研究论文,该文章提出了DeChat,这是一个基因组纠错工具,专为Nanopore R10 simplex reads 或错误率在2%左右的TGS数据设计。它利用dBG(德布鲁因图)和MSA(变异感知多序列比对)的优势,创造出一种协同方法。这种方法可避免读数过度校正,确保保留重复序列和单倍型中的变异,同时准确校正测序错误。
工具链接:https://github.com/LuoGroup2023/DeChat

原文链接:https://doi.org/10.1038/s42003-024-07376-y

研究内容总结

DeChat是专为纳米孔(ONT R10 simplex)长读长设计的重复和单倍型感知的纠错工具,仅需CPU资源,适配HAC或SUP模型的低错误率(<2%)读长数据。
DeChat具有高效处理SNP、插入和缺失等多种遗传变异,避免过度纠错的优势,并能显著降低错误率,同时保留单倍型和重复信息,防止基因组变异丢失。
DeChat与GPU依赖的深度学习工具Herro相比,DeChat更通用,避免了读长信息的大量丢失,并且在纠错效率、错误率和单倍型覆盖率方面优于其他只需CPU的工具。
使用DeChat纠错后的读数能显著提升基因组组装和宏基因组分类的质量。
DeChat方法介绍

DeChat方法分为两个阶段:
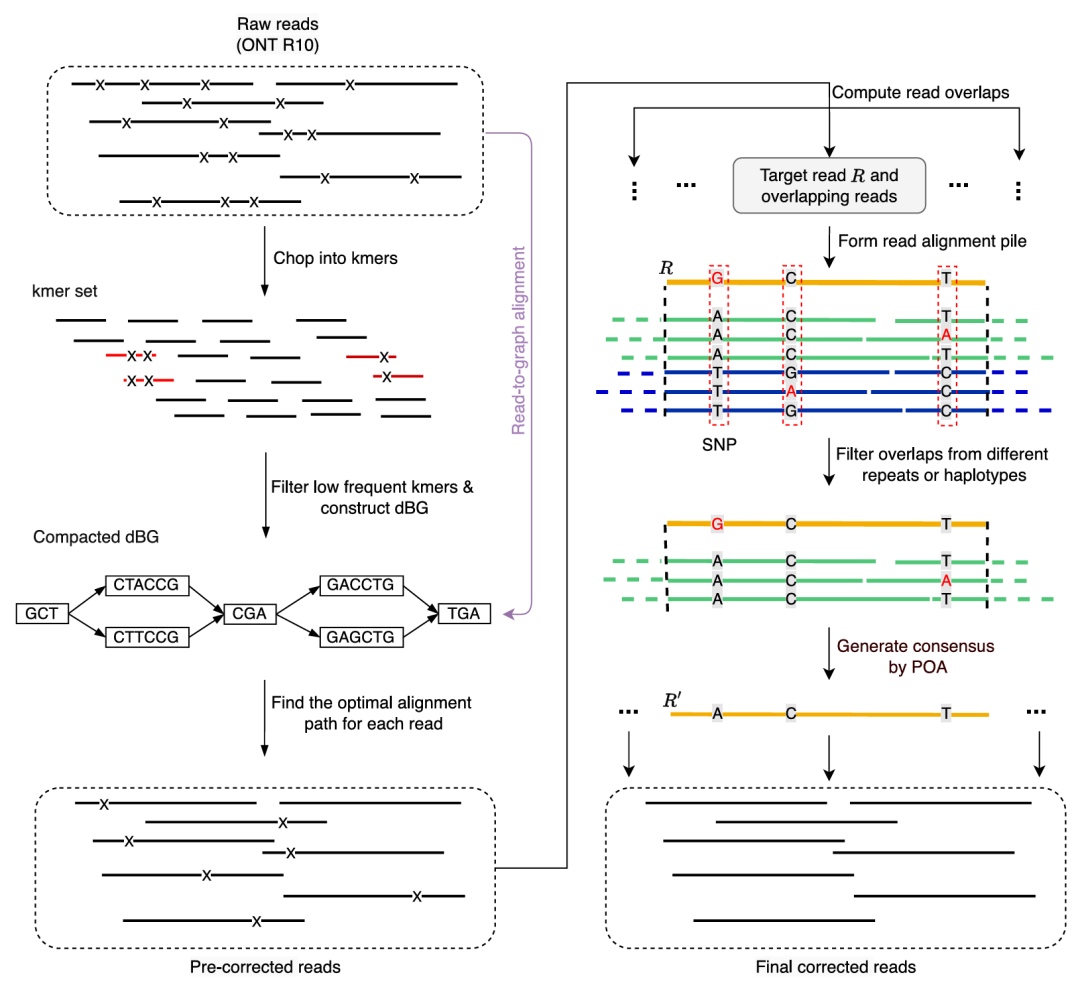
第一阶段:利用BCALM2快速构建压缩的de Bruijn图(dBG),通过最小哈希策略提高效率。随后将原始读序比对到dBG上,通过深度优先搜索和Dijkstra算法找到稳健k-mer之间的最短路径,生成预纠错的读序。
第二阶段:采用hifiasm的多序列比对模块,进行全序列比对,标记具有信息性的位点(如SNP),生成一致性序列以完成最终纠错。
如下图所示,展示了DeChat的工作流程。

模拟数据纠错
作者通过PBSIM模拟了多个ONT R10 simplex 数据集,DeChat展示了其纠错性能,在大肠杆菌多倍体(2,3,4倍体)上实现了大约 38~200、21~251 和 25~231 倍的错误率降低。在真实单倍体和四倍体马铃薯基因组上分别实现了约3.5~5.9和2~5.3倍的错误率降低。
1
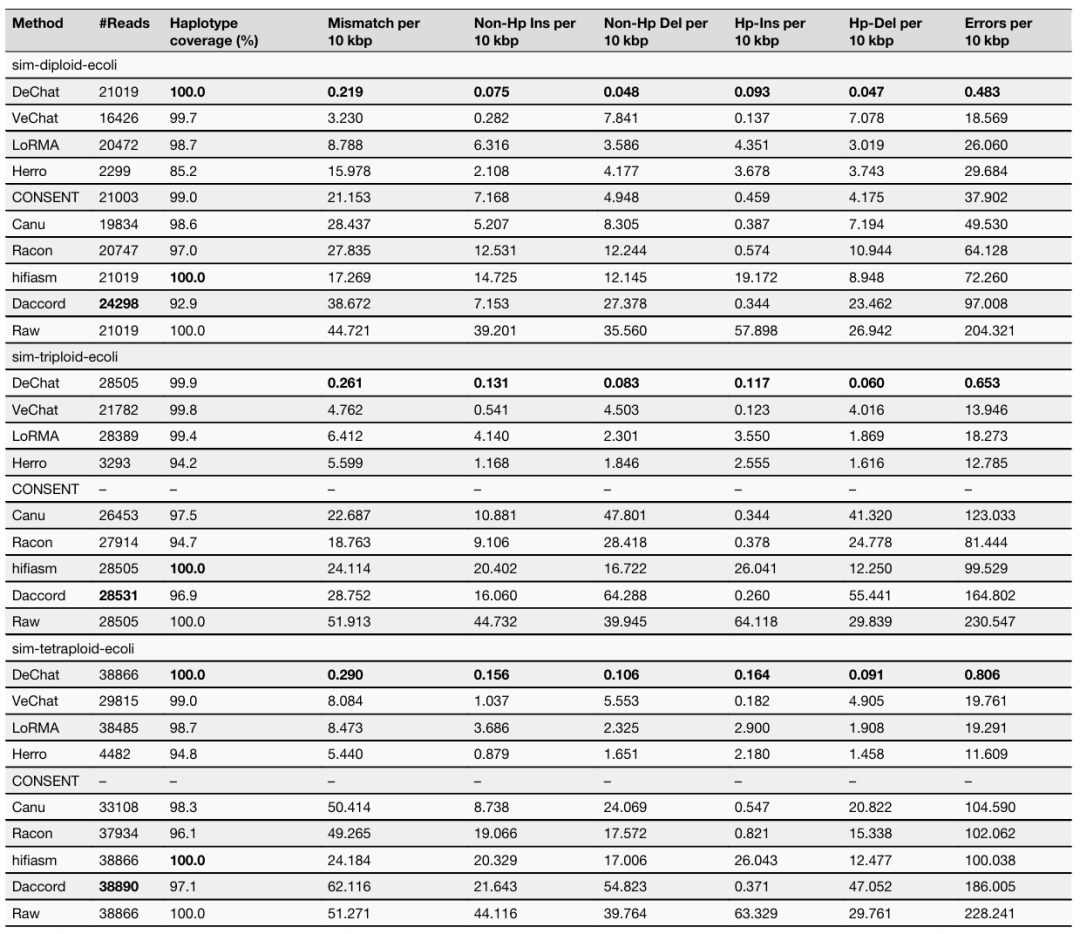
E. coli 多倍体纠错

作者通过混合大肠杆菌菌株构建了伪二倍体、三倍体和四倍体基因组,下表中展示了不同纠错工具的纠错结果。对于不同的错误类型,DeChat 在纠正Mismatch和所有四种类型的indels方面优于其他工具:non-homopolymer insertion,non-homopolymer deletion,homopolymer insertion, and homopolymer deletion。
2
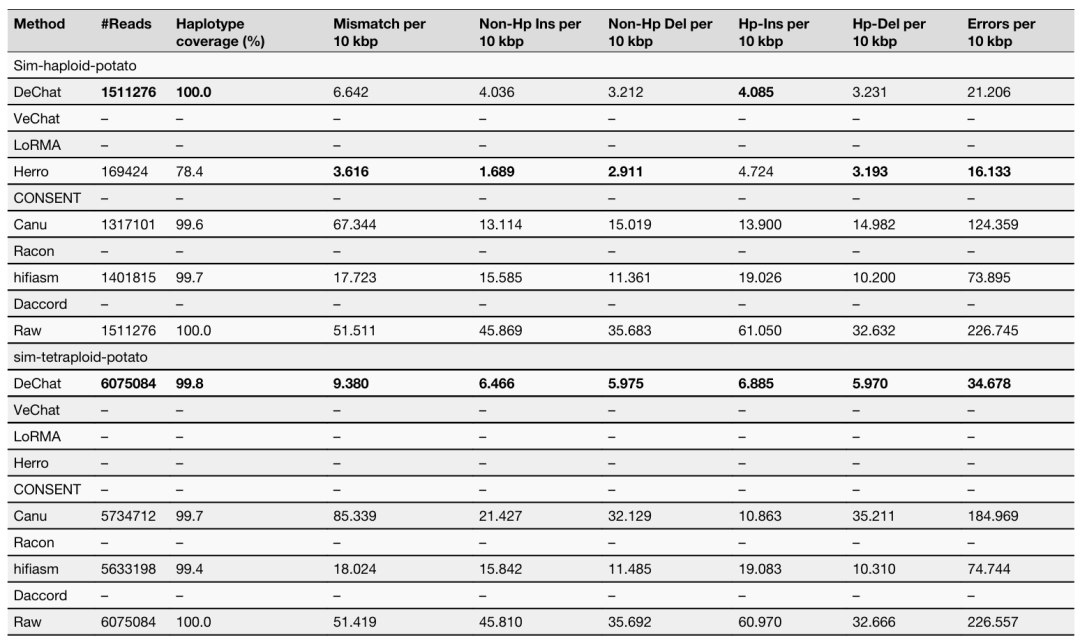
单倍体和四倍体马铃薯纠错
作者利用最近发表的高质量四倍体马铃薯基因组构建了真实的四倍体和单倍体基因组。其中马铃薯数据集中的基因组是真实的,而 ONT R10 读数是模拟合成的。“–”表示纠错工具运行错误或由于运行时间过长(CPU>2000h)而中断。
3
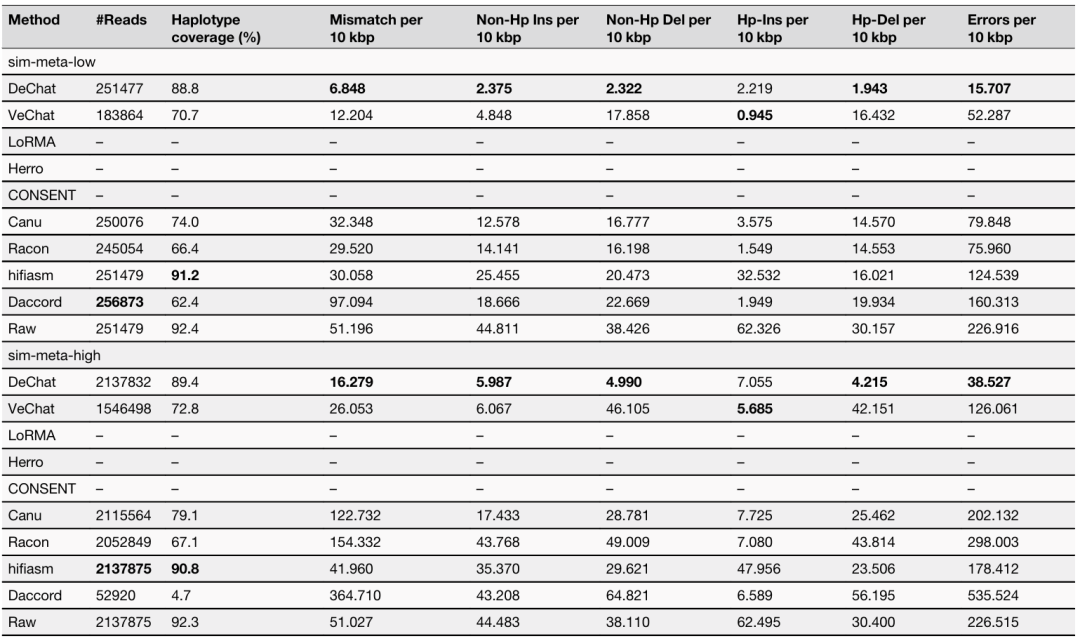
宏基因组纠错

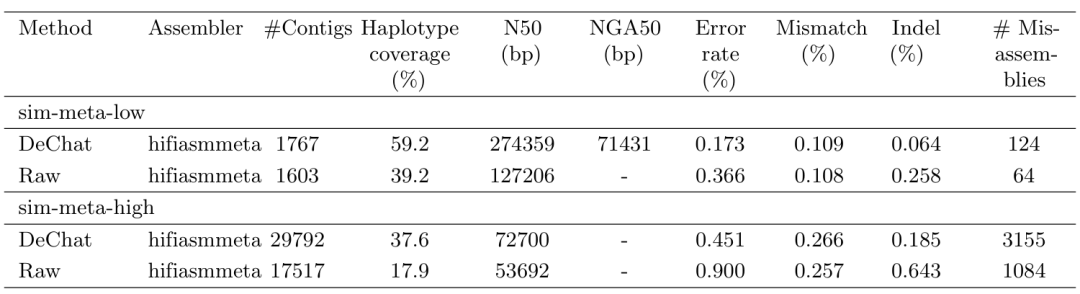
作者模拟了不同复杂度的宏基因组数据,在低复杂度(“sim-meta-low”)宏基因组和高复杂度(“sim-meta-high”)宏基因组数据集上分别将错误率降低了约 3-10 倍和 3-8 倍。CAMISIM用于模拟两个数据集,低复杂度包含 30 个物种(60 个菌株),菌株相对丰度范围为 0.30% 到 6.43%。高复杂度包含 373 个物种( 1000 个菌株),菌株相对丰度范围为 0.04% 至 0.30%。两个数据集中涉及的微生物基因组均来自于 RefSeq 中存放的完整基因组序列。

真实数据纠错

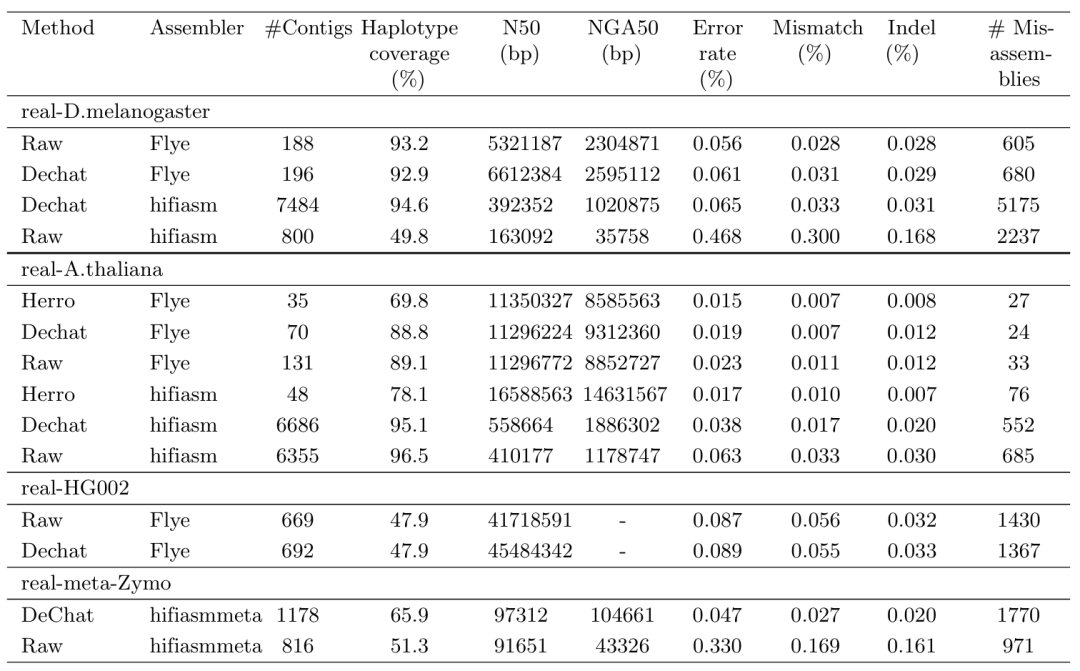
作者还针对果蝇和拟南芥以及宏基因组(real-meta-Zymo) 的ONT R10 simplex reads 测试了各种纠错方法的性能,与大多数其他方法相比,真实 D.melanogaster 数据集的错误率低 1.2 到 1.4 倍,真实拟南芥数据集的错误率低 1.2 到 1.4 倍。虽然DeChat 在拟南芥数据集上的错误率高于 Herro,但 Herro纠错后丢失了约 90%的reads,这显着影响单倍型覆盖率,使其减少约 15%。对于真实宏基因组zymo,DeChat与单倍型覆盖率相似的其他方法(例如 ashifiasman 和 Racon)相比,DeChat 分别显着降低了大约 2 倍和 5 倍的错误率。而其他擅长降低错误率的工具却在降低错误率的同时,使大量信息丢失,单倍型覆盖率下降了10%至15%。
1
果蝇和拟南芥纠错
real-D.melanogaster 的平均测序覆盖率为 129层,real-A.thaliana 的平均测序覆盖率为 223层。“–”表示纠错工具运行错误或由于运行时间过长(CPU>2000h)而中断。
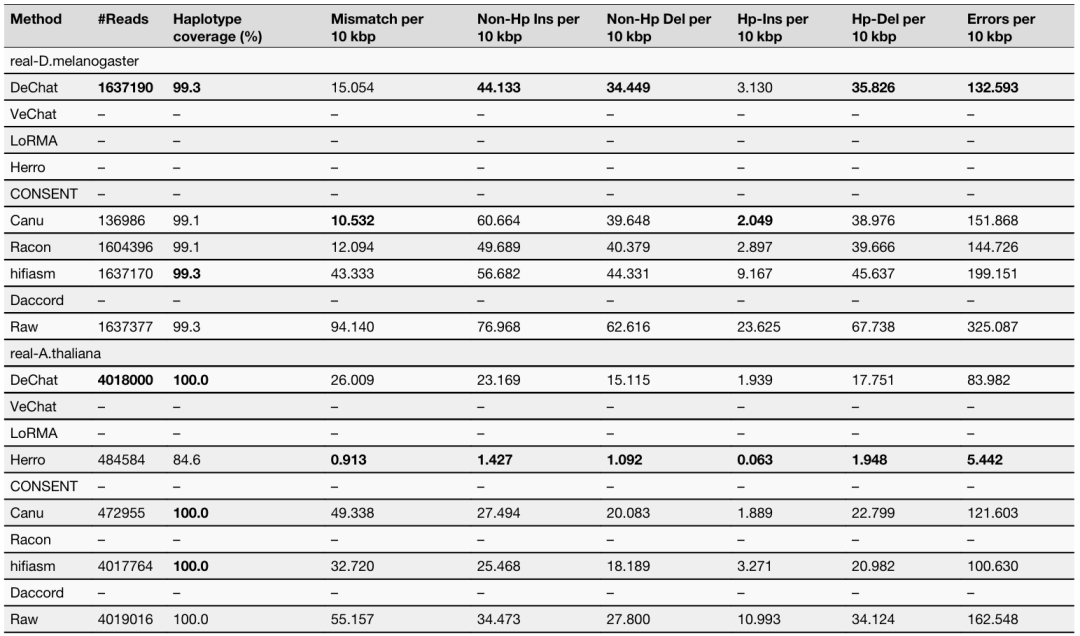
2
宏基因组zymo纠错
zymo数据集测序错误率约为 2%,平均测序覆盖度是 30 层。“–”表示纠错工具运行错误或由于运行时间过长(CPU>2000h)而中断。
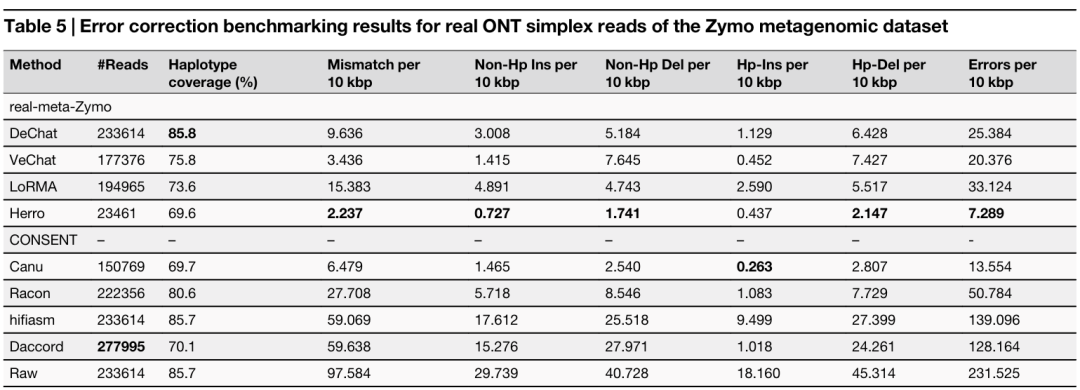
3
单倍体马铃薯重复区域纠错

作者从单倍体马铃薯基因组的重复区域中提取了校正(子)读数,使用 EDTA生成了这些重复区域的坐标,结果显示,DeChat 在错误率(mismatch和indels)和马铃薯基因组重复区域内的单倍型覆盖度方面均优于其他可用工具。
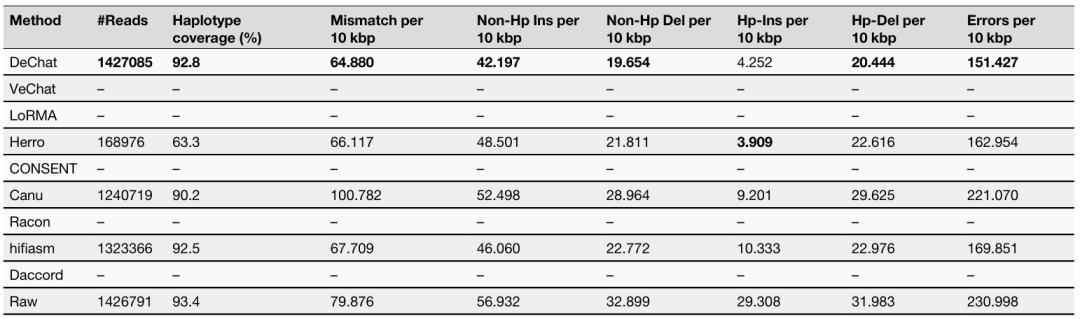
除此之外,作者还针对模拟的大肠杆菌和马铃薯数据,分别针对读数在不同长度、覆盖度以及错误率的情况下的评估,均显示了DeChat在纠错结果各方面的优越性。


组装与分类改进

为了研究纠正错误后的读数对新基因组组装质量的影响,作者分别使用 hifiasm 和 hifiasm-meta(针对多倍体基因组和宏基因组的两种组装程序)开展了一系列实验,并在宏基因组测序数据上对分类性能进行了比较研究。
1
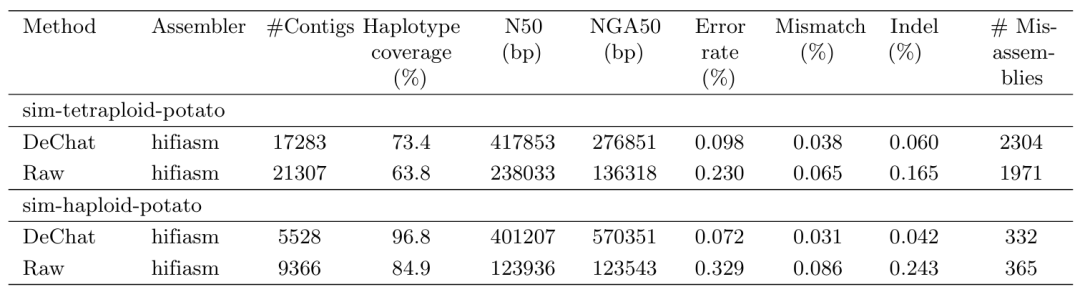
模拟数据集的组装评估
在下方所有数据集中,在组装之前应用 DeChat 可以显着提高单倍型覆盖度、获得更长的contig(通过 N50/NGA50 测量)和更低的错误率。
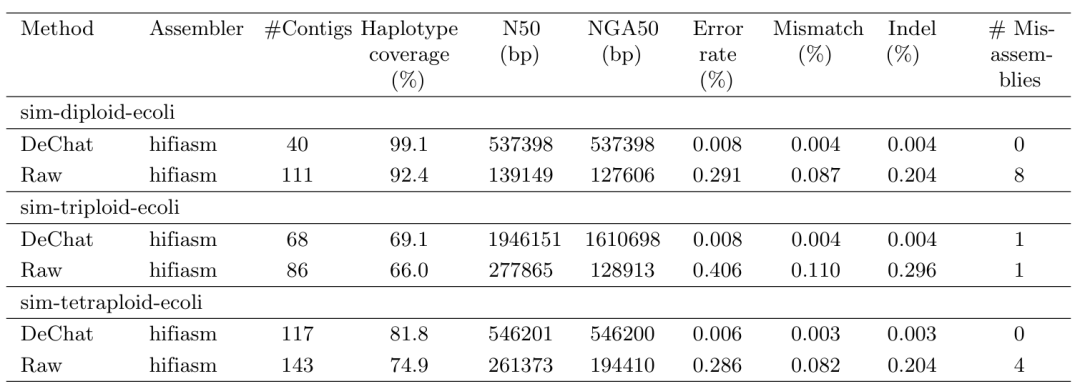
2
真实数据集的组装评估
即使是对于纠错后的ONT reads,还是会产生较低的错误率,而 hifiasm 通常会产生具有重复序列的较大的 contig ,导致 N50 低于 NGA50 。为了解决这个问题,作者还应用了Flye组装工具。DeChat+Flye 的组合显着提高了基因组组装的连续性 (N50/NGA50),同时在其他指标上保持与原始 Flye 组装相当的性能。
3
单倍体马铃薯重复区域纠错

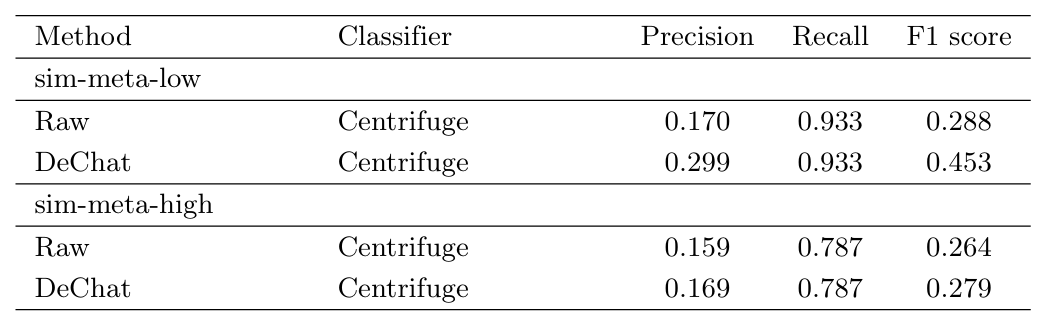
作者还针对宏基因组专门评估了纠错后的分类结果。如下表所示,DeChat + Centrifuge 的组合在保持相同 Recall 的同时显着提高了 Precision 。
感谢阅读!
欢迎批评指正。
撰文|李毅晨
内容中包含的图片若涉及版权问题,请及时与我们联系删除

评论
沙发等你来抢