


论文题目:Enabling Fast, Accurate, and Efficient Real-Time Genome Analysis via New Algorithms and Techniques
作者:Can Firtina
类型:2024年博士论文
学校:ETH Zurich(瑞士苏黎世联邦理工学院)
下载链接:
链接: https://pan.baidu.com/s/1wVi5V7VQXb45Dadp572lNw?pwd=n1qi
硕博论文汇总:
链接: https://pan.baidu.com/s/1Gv3R58pgUfHPu4PYFhCSJw?pwd=svp5
基因组分析在个性化医疗 [2–22]、基因组编辑 [23–50]、进化生物学 [51–65]、癌症研究 [66–112]、产前和新生儿筛查 [113–137]、疫情追踪 [138–157]、微生物组研究 [158–182]、农业 [183–195] 和法医学 [196–214] 等各个领域发挥着至关重要的作用。高通量测序 (HTS) 技术的出现,例如边合成边测序 (SBS) [215–223]、单分子实时 (SMRT) [224] 和纳米孔测序 [225–247],彻底改变了基因组分析,与桑格测序 [249] 相比,它能够以相对较低的成本生成大量基因组数据,从而实现更快、更具成本效益的基因组测序 [248]。然而,由于多种原因,基因组数据分析具有挑战性:1)HTS 技术只能对相对较短的基因组片段(称为读段)进行测序,而这些片段在相应基因组中的位置未知 [250–254];2)这些读段可能包含测序错误 [248, 252, 255–257],导致与原始序列不同;3)由于物种内和物种间个体之间的差异,测序的基因组可能(通常不会)与参考数据库中记录的基因组(称为参考基因组)不完全匹配 [255,258]。尽管自 20 世纪 80 年代以来,计算工具取得了显着改进 [258],以克服这些挑战,但基因组数据的快速增长 [259] 导致基因组分析流程中的计算开销越来越大,对高效、准确和及时的基因组分析提出了巨大挑战 [260–262]。
图 1.1 显示了基因组分析流程的多个关键步骤,每个步骤都会影响基因组分析的准确性、速度和能耗。
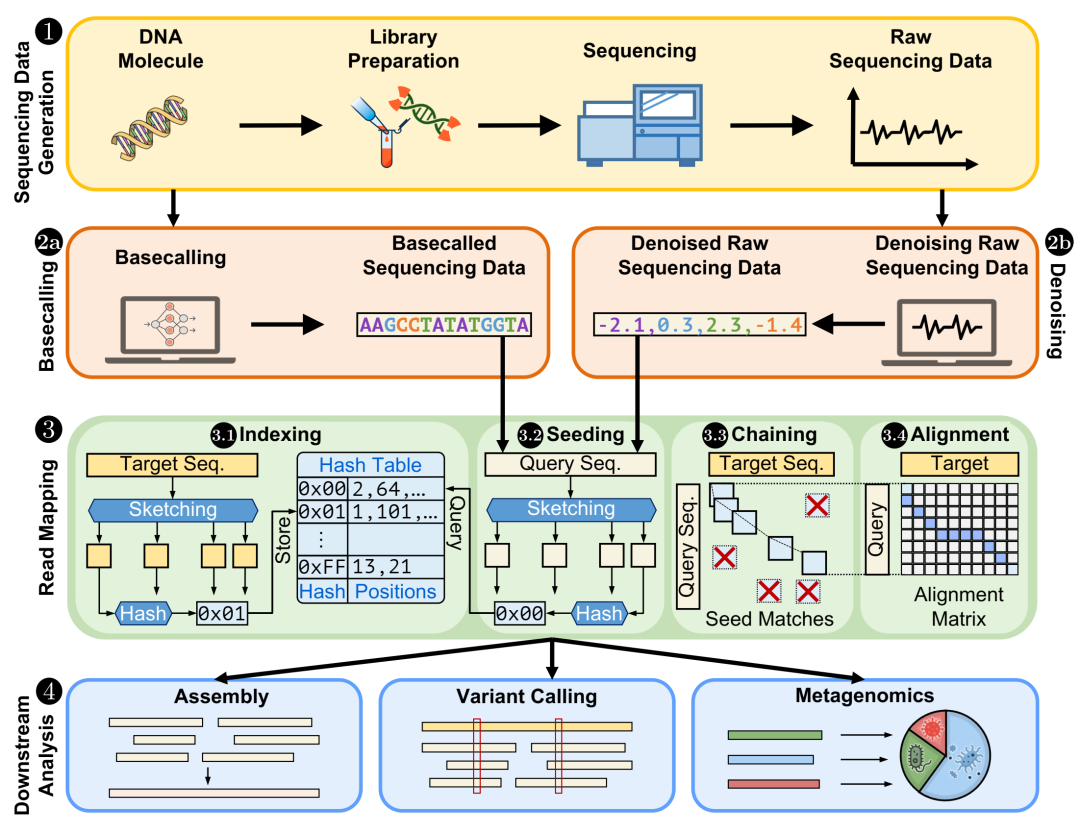
第三,读取映射 3 旨在查找基因组序列对之间的相似性和差异(例如,一个或多个物种的测序查询读取和目标参考基因组之间的相似性和差异)。为了便于实际的相似性识别,读取映射包括几个步骤。这些步骤包括构建(即索引 3.1 )和使用(即播种 3.2 )数据库 [2, 265, 266, 292,299, 304–411],通过利用各种草图(即采样)[306,310, 333, 360, 412–432] 和散列 [304, 307–309, 315, 334, 358, 414, 416, 424, 433–459] 方法进行有效的相似性搜索。读取映射中的过滤 [357, 460–468] 和共线链接(即稀疏动态规划)[306, 469–482] 步骤 3.3 旨在通过快速识别查询和目标序列之间高度不同或相似的区域来减少读取映射中接下来计算成本高昂的步骤的工作量。比对 [483–501] 步骤 3.4 识别查询和目标序列之间的确切差异和相似性,由于基因组序列的规模很大,因此总体上需要相当大的处理能力和内存 [258, 502, 503]。第 2.2 节提供了读取映射中关键步骤的详细背景。
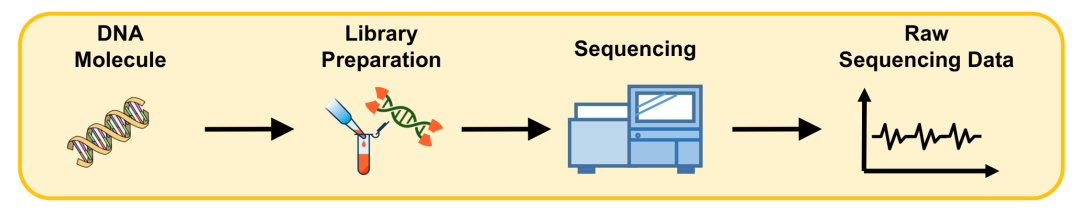
测序数据生成的主要步骤。
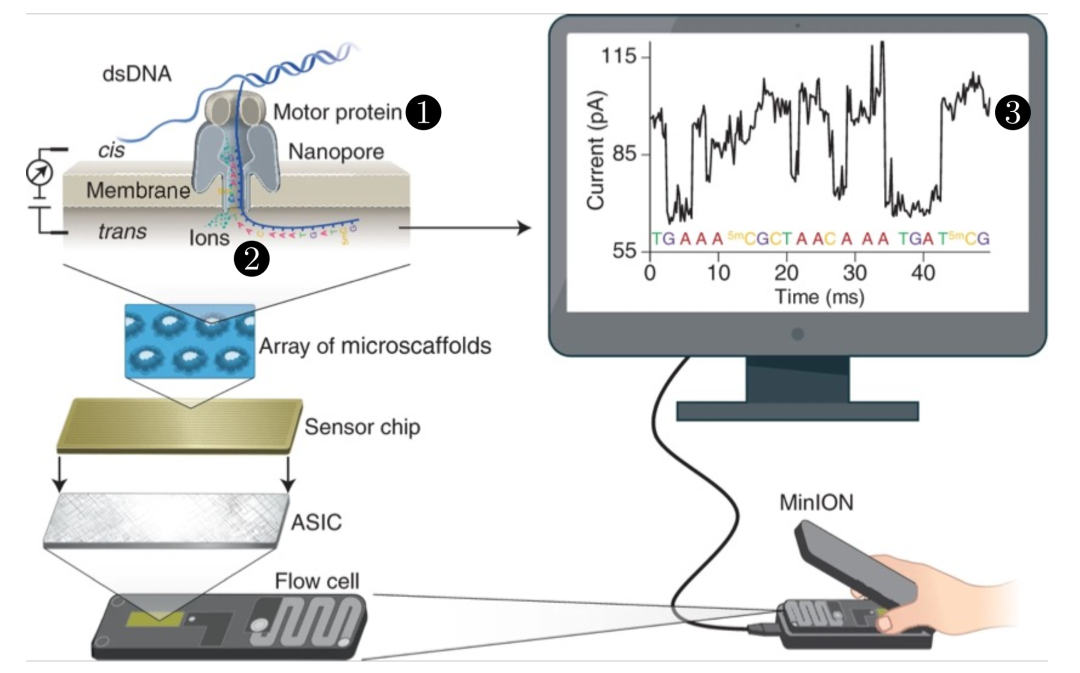
纳米孔测序仪的结构及其测序步骤。
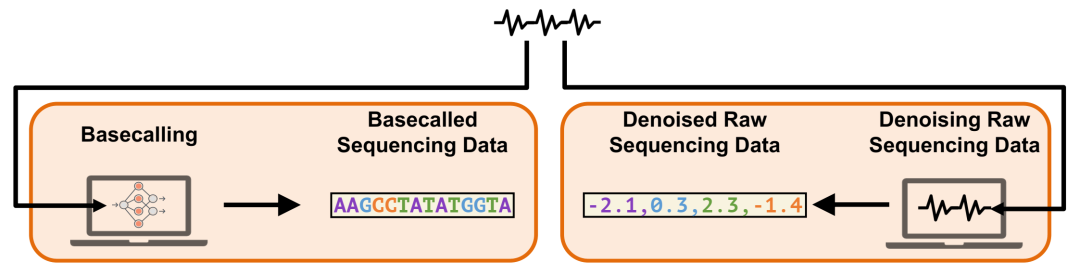
处理原始测序数据的主要步骤。
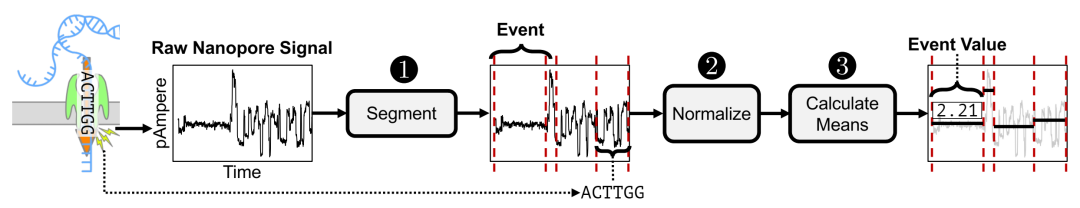
原始信号分析的主要步骤。
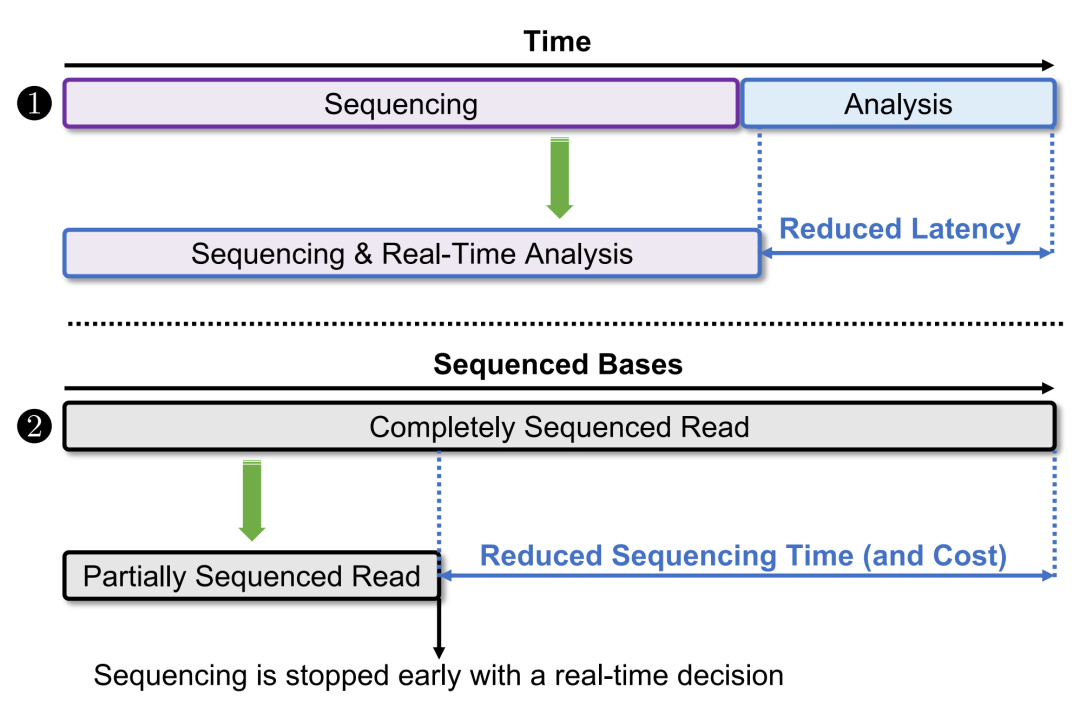
纳米孔测序实时分析的两个主要优点。
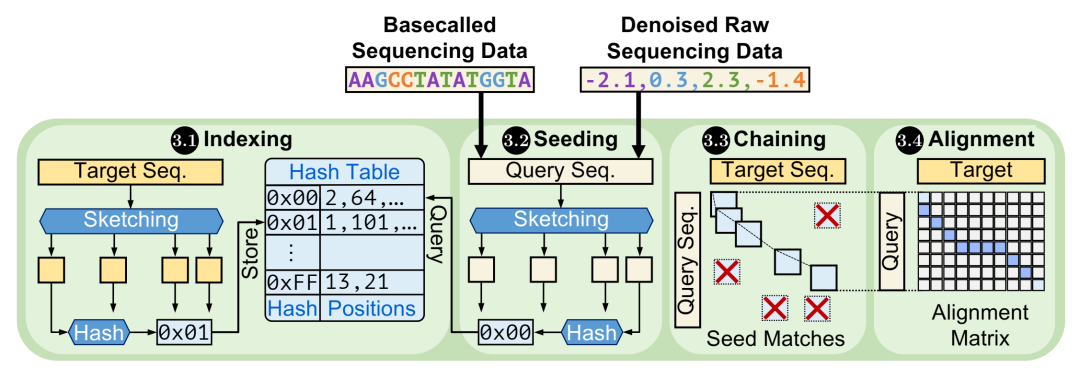
读取映射的主要步骤。
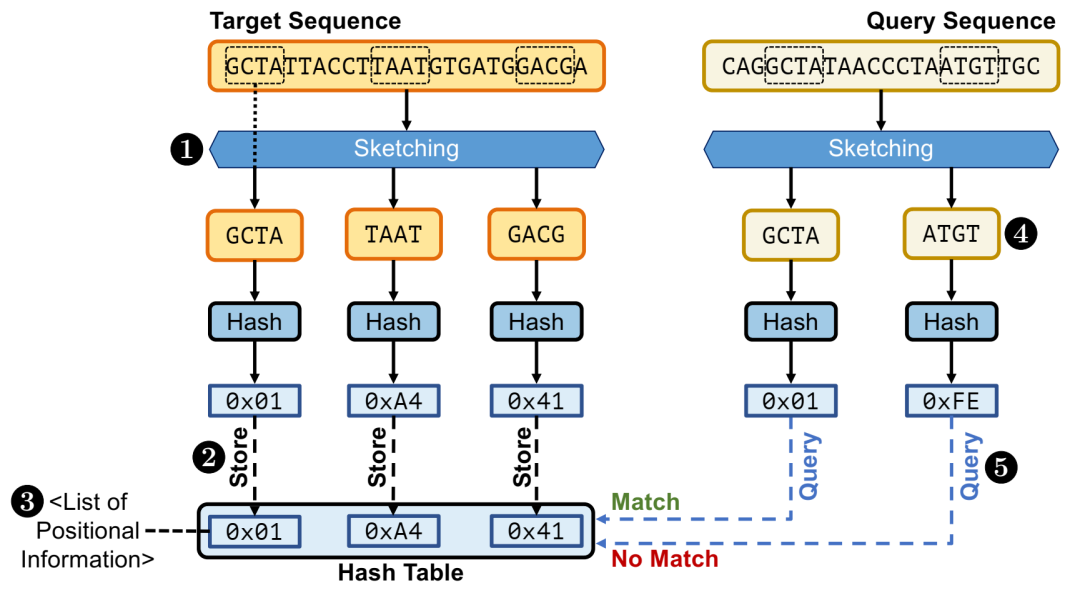
索引(左侧)和种子(右侧)的步骤,使用哈希值在目标序列和查询序列之间找到匹配的序列段。
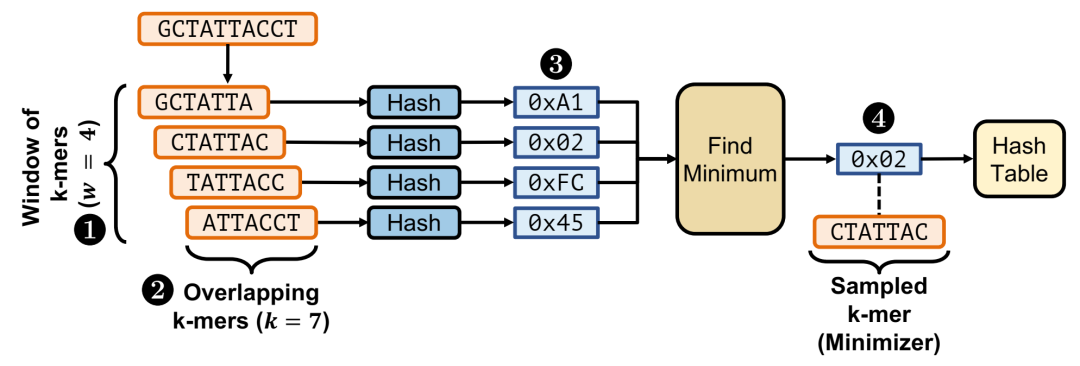
一种采样(即草图)机制,称为最小化草图。
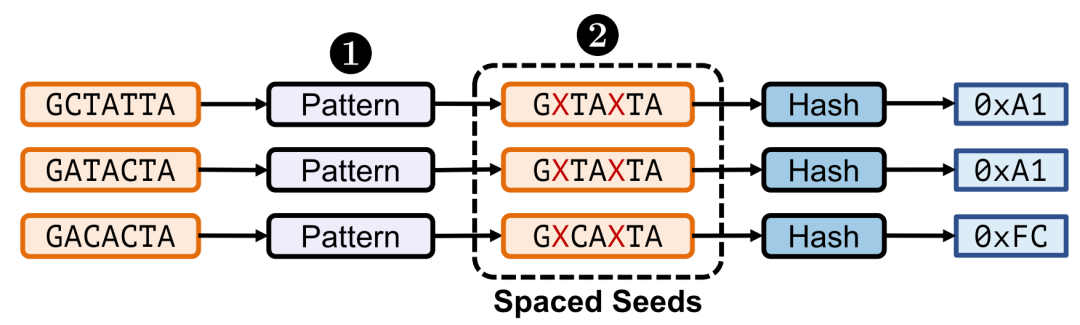
间隔播种技术。预定义的固定模式应用于所有三个输入序列。屏蔽字符以红色 X 突出显示。前两个输入序列生成相同的输出哈希值,因为它们彼此不同的字符被屏蔽,从而生成相同的间隔种子和这些种子的相同对应哈希值。
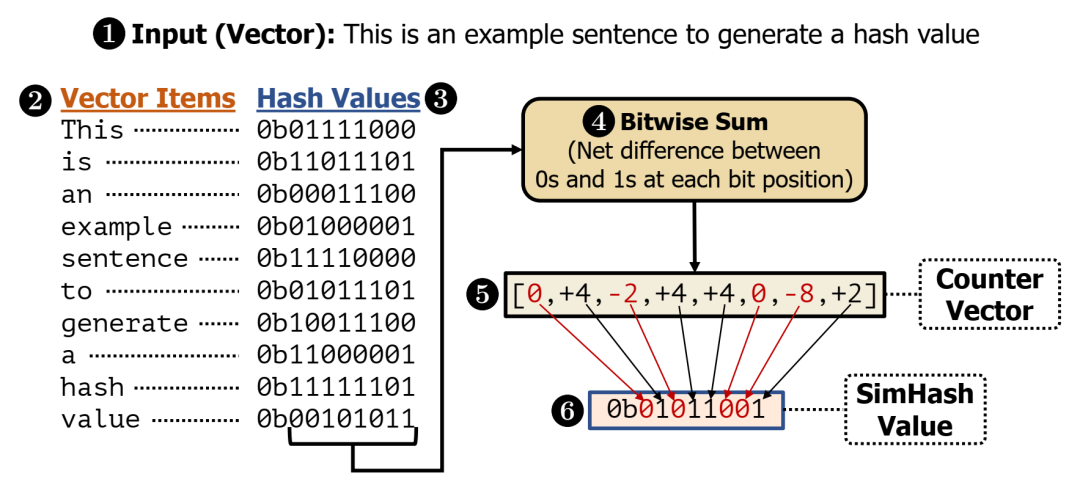
使用 SimHash 技术为项目向量生成哈希值。示例输入是一个句子(即向量),其中这些单词(即项目)的哈希值(以二进制形式显示)用于生成整个句子的哈希值。
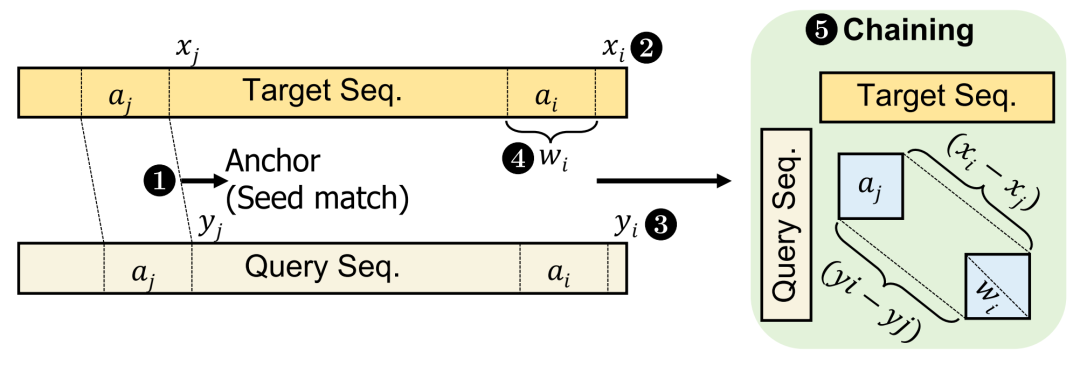
在目标序列和查询序列之间链接锚点(种子匹配)。链接方法计算一对锚点之间的距离,以确定它们是否应包含在同一个链中。
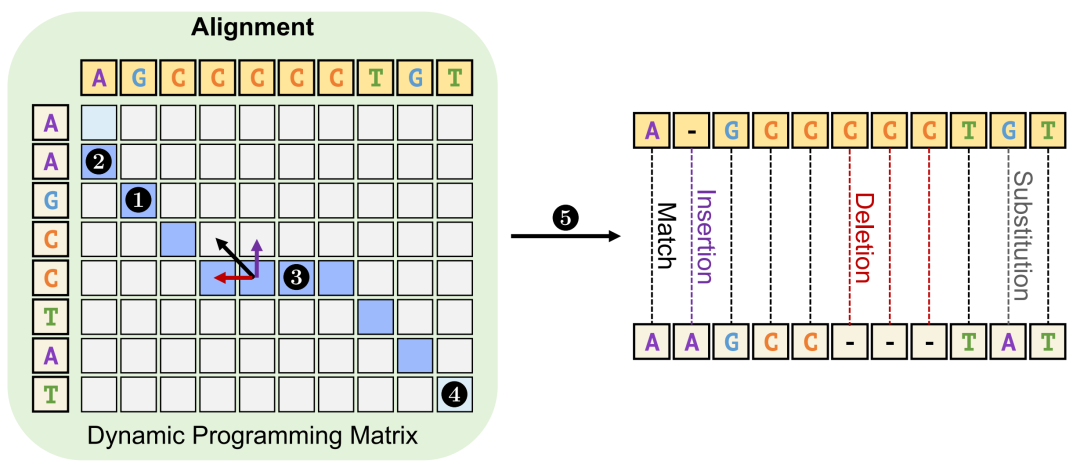
动态规划 (DP) 矩阵用于识别编辑操作:匹配、替换、插入和删除。每个单元格的值都是根据相邻单元格的值计算的,如箭头所示。最佳对齐路径以深蓝色突出显示,而浅蓝色单元格表示在链接步骤中识别的两个后续锚点。最佳对齐的相应编辑操作显示在 DP 矩阵的右侧。
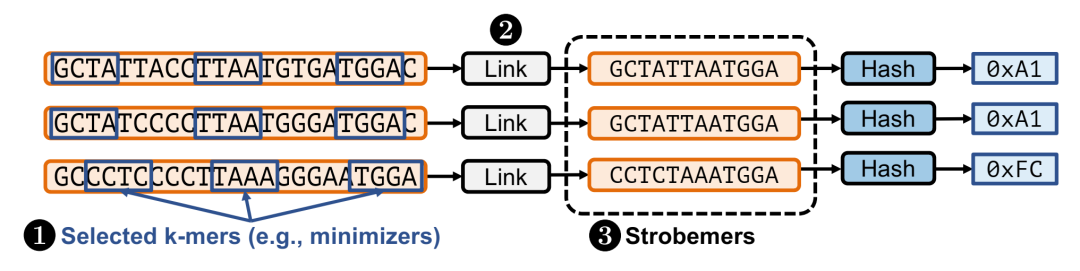
具有三个不同输入序列的频闪器技术的简单示例。所选 k-mer 之间的序列被忽略,以容忍插入和删除。前两个输入序列在生成频闪器种子后,会生成与输出相同的哈希值,即使这些输入序列并不完全匹配。
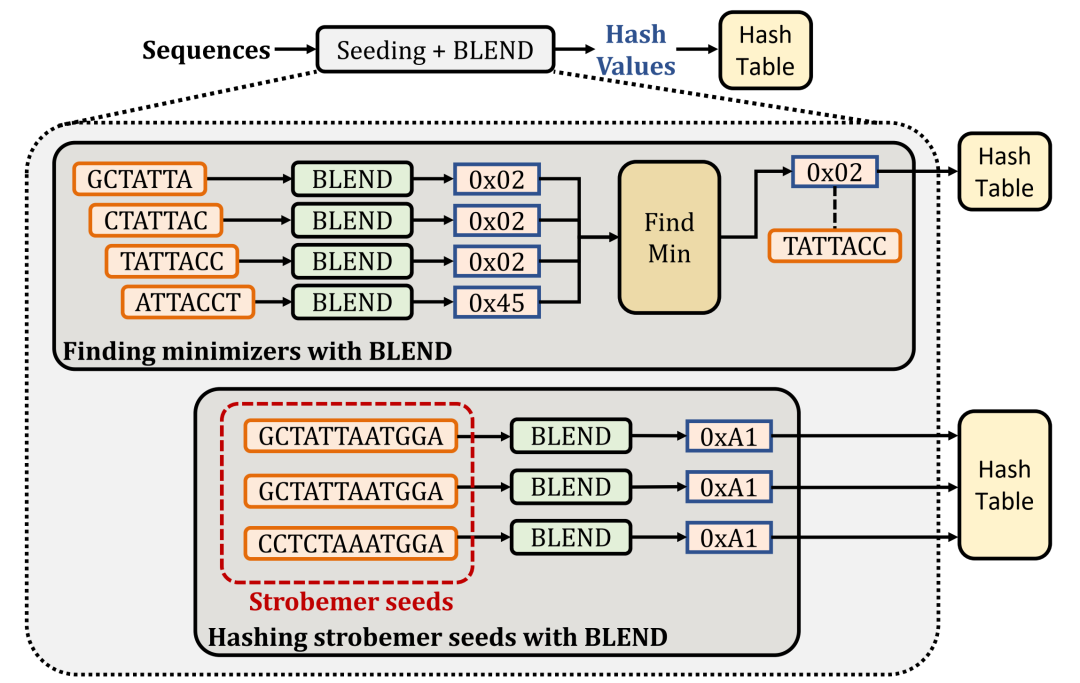
用 BLEND 替换种子技术中的哈希函数。
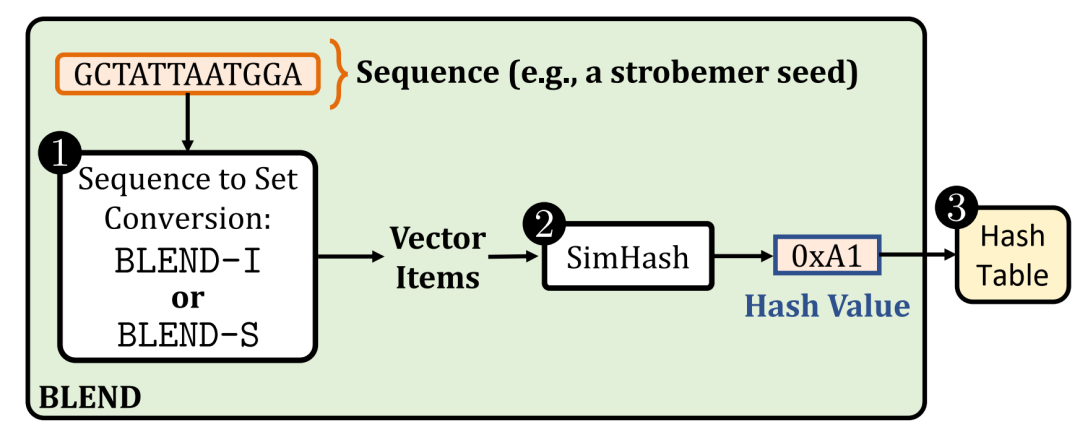
BLEND 概述。1 BLEND 使用 BLEND-I 或 BLEND-S 将序列转换为其项目向量。2 BLEND 使用其项目向量和 SimHash 技术生成输入序列的哈希值。3 BLEND 使用哈希表查找模糊种子匹配,只需查找 BLEND 生成的哈希值即可。
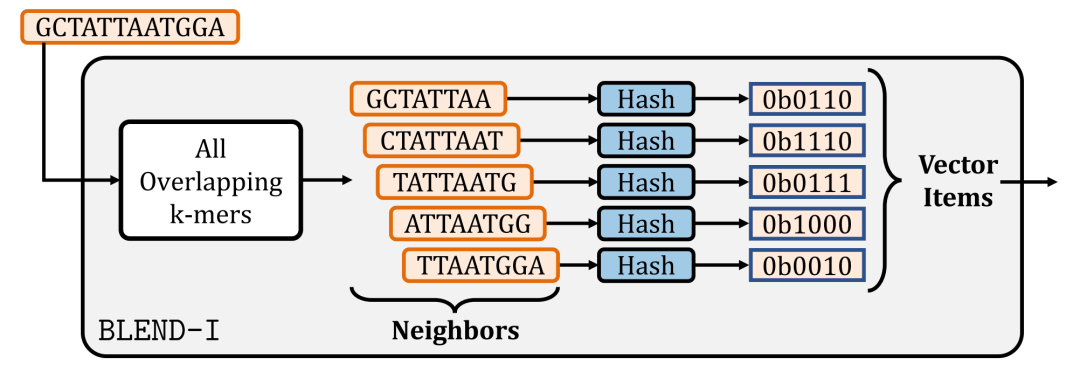
BLEND-I 概述。BLEND-I 使用输入序列所有重叠 k-mer 的哈希值作为向量项。

BLEND-S 概述。BLEND-S 仅使用由频闪序列播种机制选择的 k-mer 的哈希值。
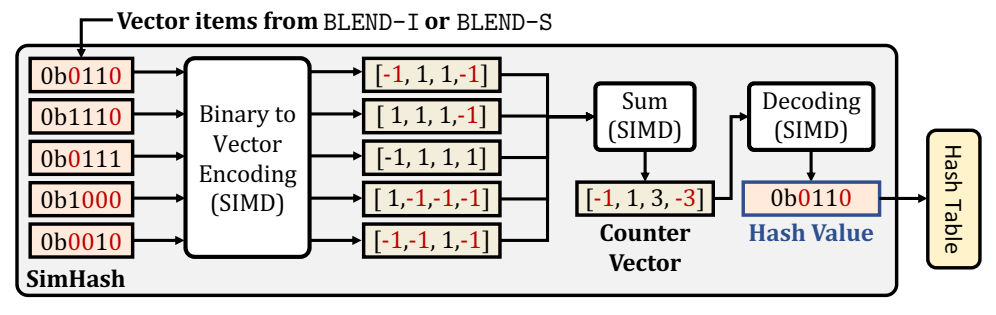
SimHash 技术中用于计算给定项向量的哈希值的步骤概述。向量项是以二进制形式表示的哈希值。二进制到向量编码将这些向量项转换为其对应的位向量表示。Sum 执行向量加法并将结果存储在单独的向量中,我们将其称为计数器向量。解码根据计数器向量中的值生成向量的哈希值。BLEND 使用 SIMD 操作执行这三个步骤,如 SIMD 所示。我们用红色突出显示了 0 位在 SimHash 技术中的转换和传播方式。
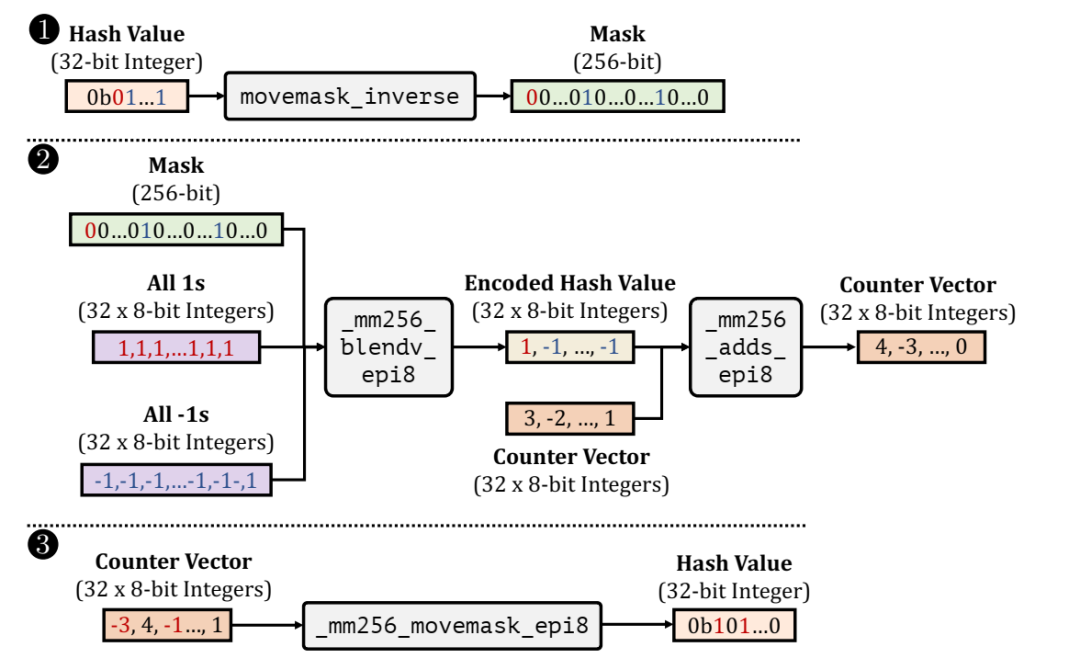
从 BLEND-I 或 BLEND-S 识别的向量项生成种子的哈希值时的 SIMD 执行流程。颜色突出显示位和值传播到函数的输出。
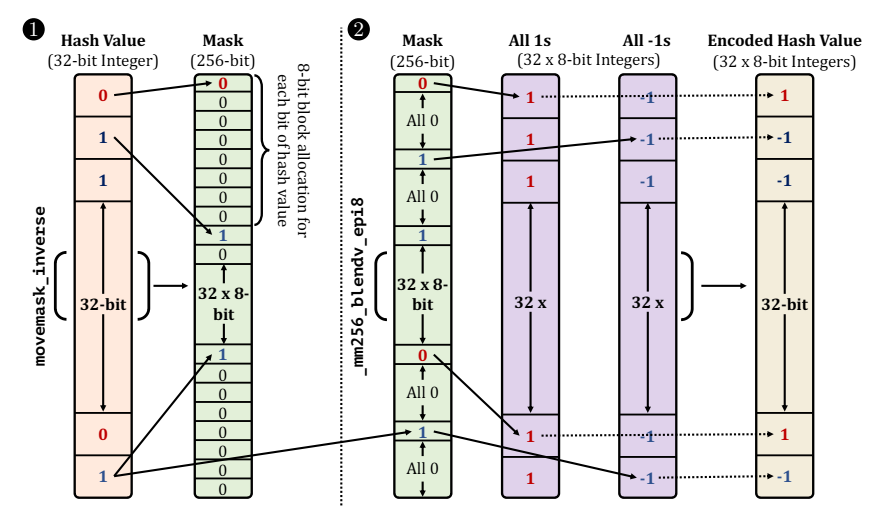
movemask_inverse 和 _mm256_blendv_epi8 执行的详细信息。颜色和箭头突出显示位和值到函数输出的传播。

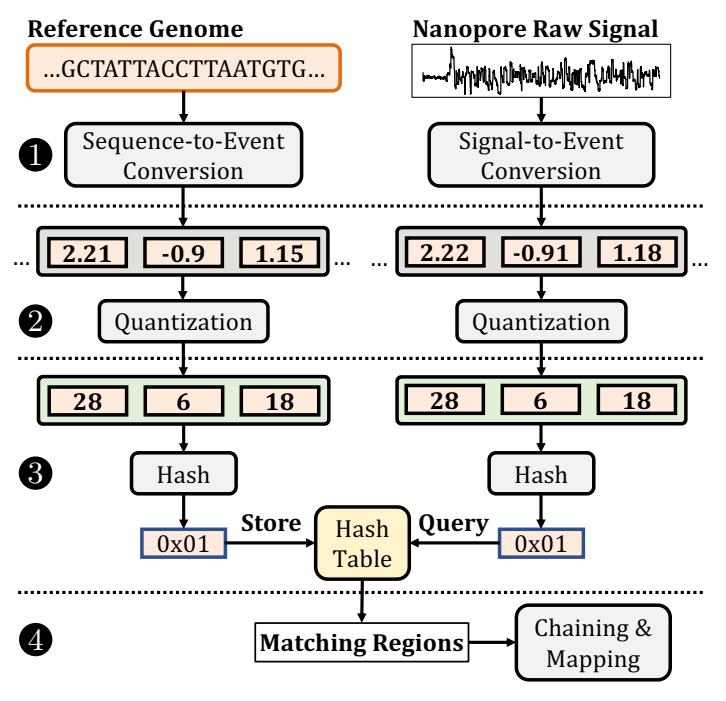
RawHash 概述。
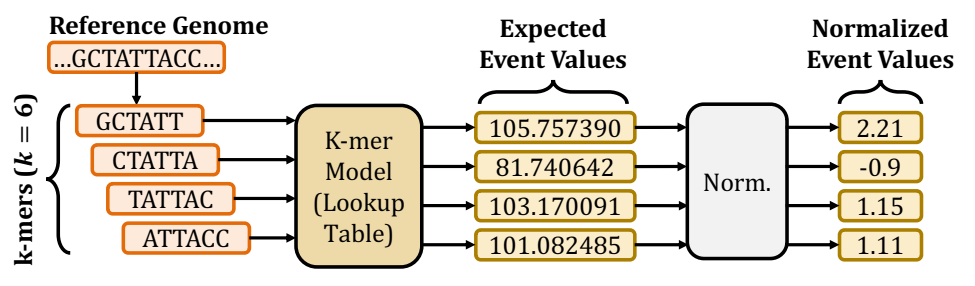
根据纳米孔的 k-mer 模型将序列转换为事件值。

从原始信号中检测事件。
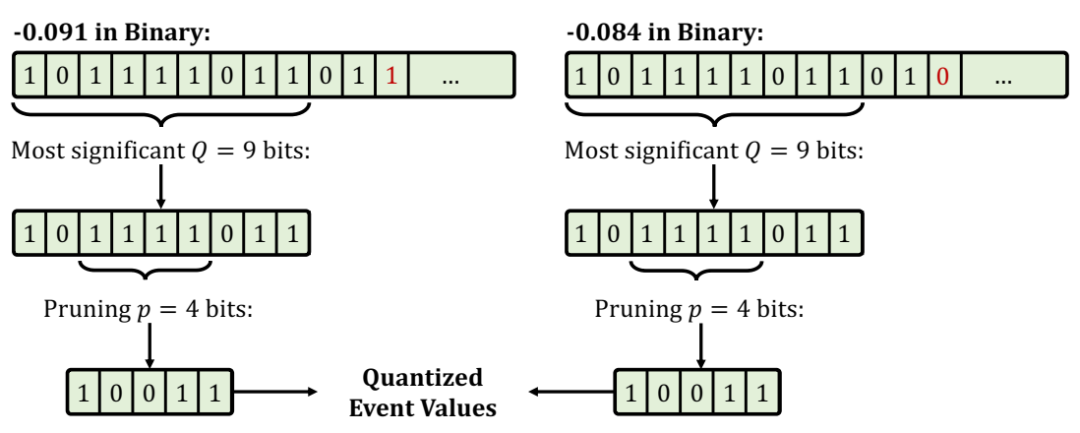
两个事件值的量化。
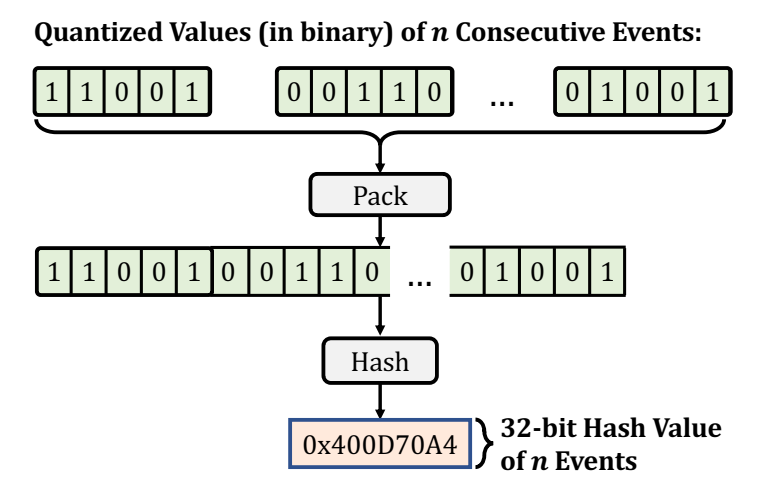
从 n 个连续的量化事件值生成一个哈希值。
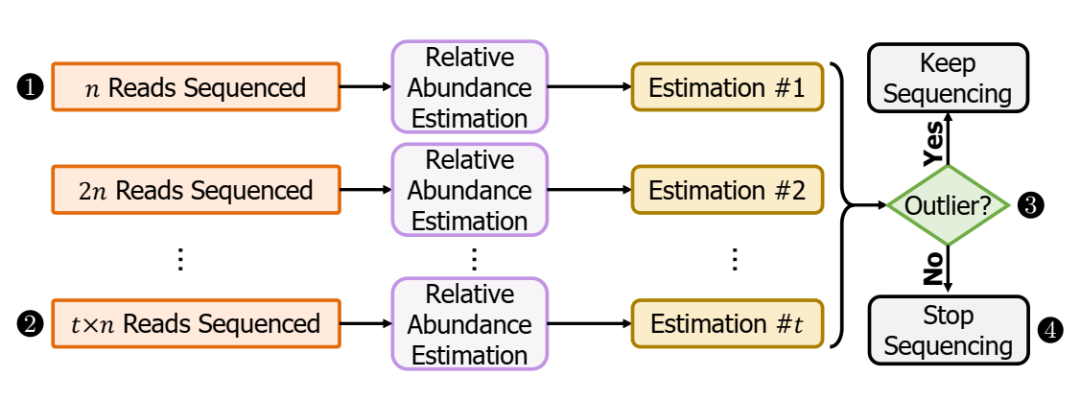
Sequence Until 机制的概述。
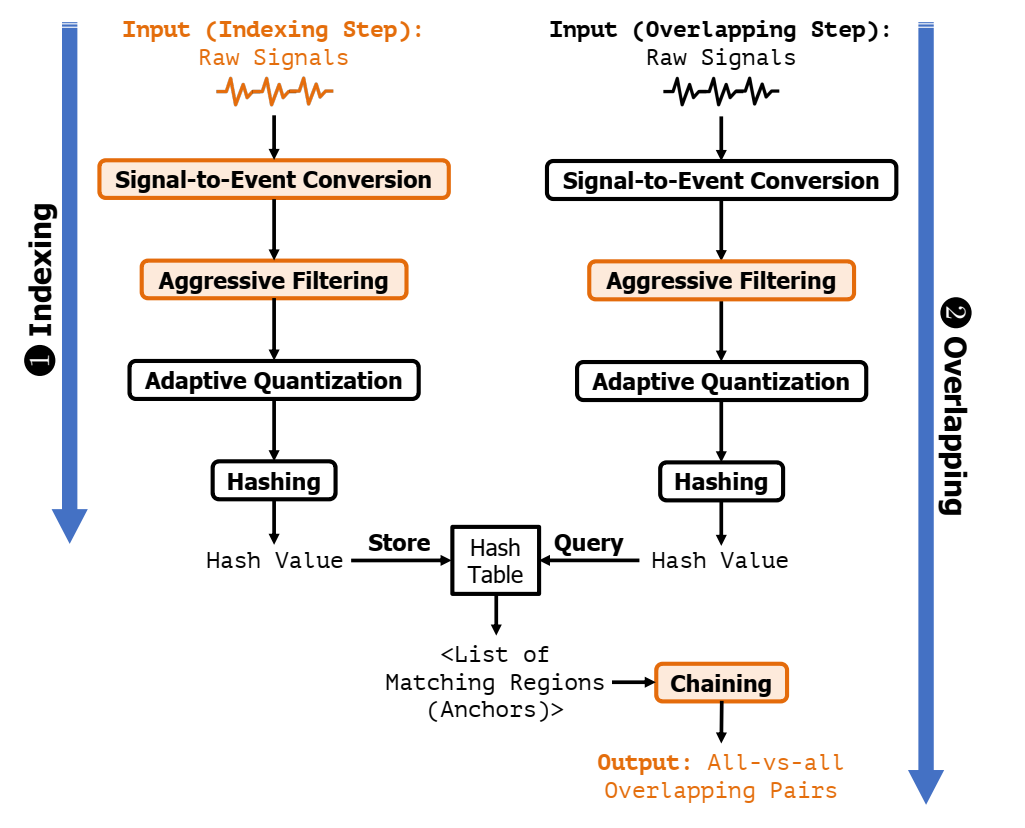
Rawsamble 概述。我们使用颜色来表示输入、步骤和输出,以突出显示 Rawsamble 在 RawHash2 上修改的部分。
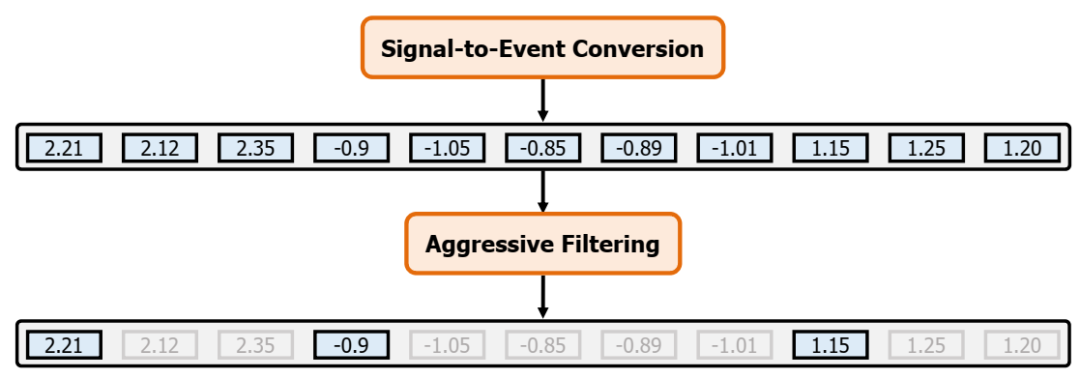
在 Rawsamble 中进行过滤。灰色框中的值显示已过滤的信号,因为它们的值接近之前未过滤掉的信号(蓝色框中)。
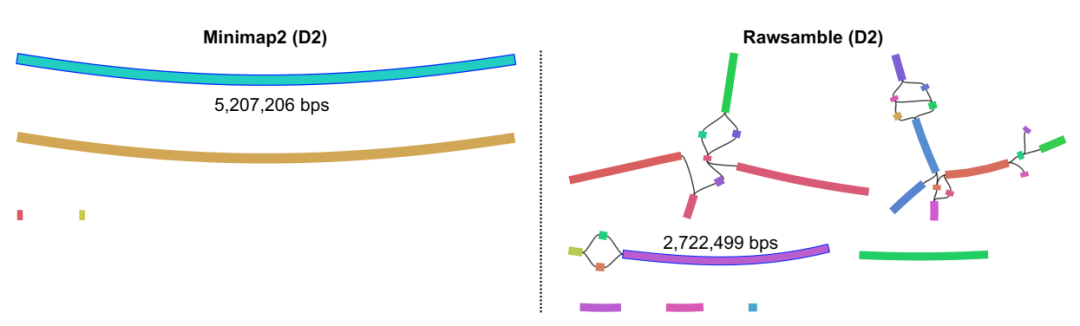
使用 Rawsamble 和 minimap2 生成的组装图可视化与 D2(大肠杆菌)数据集重叠。

使用 Rawsamble 和 minimap2 生成的组装图可视化与 D3(酵母)数据集重叠。
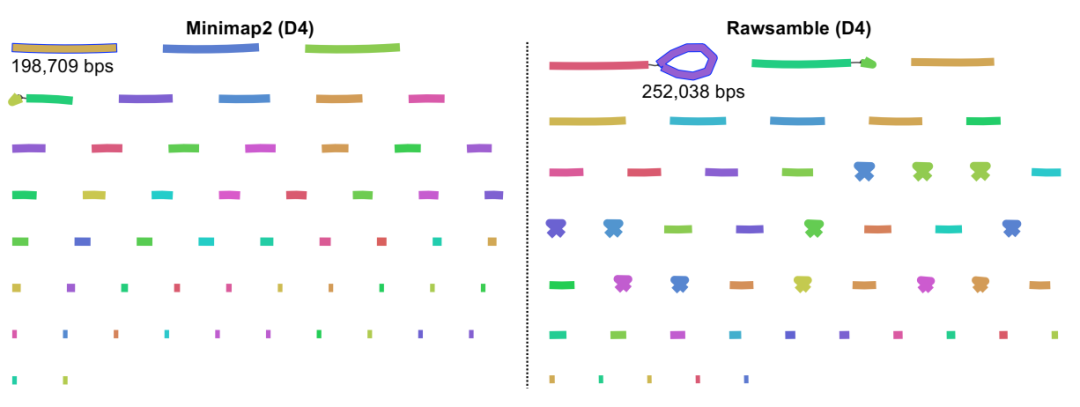
使用 Rawsamble 和 minimap2 生成的组装图可视化与 D4(绿藻)数据集重叠。
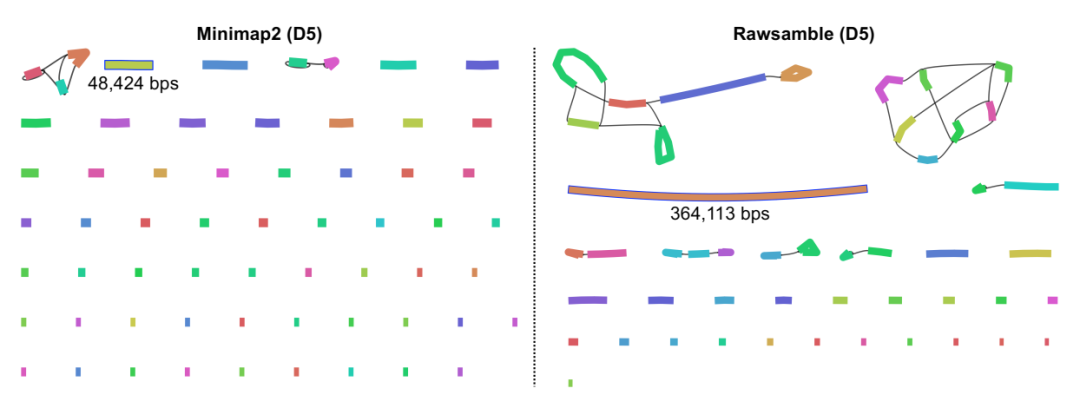
使用 Rawsamble 和 minimap2 生成的组装图可视化与 D5(人类)数据集重叠。







内容中包含的图片若涉及版权问题,请及时与我们联系删除

评论
沙发等你来抢