
DRUGAI
目前,大多数应用于凝聚态系统的机器学习势(machine learning potentials, MLPs)主要依赖于多体置换不变多项式(many-body permutationally invariant polynomials)或以原子为中心的神经网络(atom-centered neural networks)。然而,这些方法在实现原子级能量分解的化学可解释性方面面临挑战,同时也难以完全匹配传统力场的计算效率。本研究中,研究人员提出了一种融合上述两类方法优势的新策略,在准确性与接近力场级速度之间取得了良好平衡。该方法采用以单体为中心的表示方式(monomer-centered representation),将总势能分解为一系列具有化学意义的单体能量之和。每个单体的结构描述符由一体项和二体有效相互作用组成,这些相互作用通过适当构建的置换不变多项式输入至前馈神经网络中,从而实现能量预测。 研究人员对该方法在不同系统中的表现进行了系统评估,包括气相水三聚体、液态水、甲烷-水簇以及液态二氧化碳等体系。结果表明,该方法在准确性、计算效率和灵活性方面均有所提升,显示出在构建高精度机器学习势方面的巨大潜力,并有望应用于复杂分子系统的大规模量子力学与经典力场模拟中。

分子体系的计算模拟对于理解化学、生物学和材料科学中的复杂过程至关重要。然而,在量子或经典模拟中,势能与力的评估往往计算代价极高。尽管如CCSD(T)等高精度电子结构方法被视为理想手段,但其计算量在体系原子数超过15时便变得难以承受。密度泛函理论虽然相对高效,因此常用于从头分子动力学模拟,但其精度有限且计算扩展性仍不理想,难以支持大体系的长时间模拟。
过去二十年,机器学习势(MLPs)逐渐成为一种可行的高效且准确的模拟手段。在处理上万个原子的高维体系时,原子级势能分解是一种常用策略,即将总势能表示为所有原子的局部能量之和。该方法广泛应用于多种主流MLPs模型中。但类似于电子结构理论中原子轨道与分子轨道能量的区别,原子局部能量缺乏明确的化学意义,且其物理定义不清,可能导致神经网络产生任意分配,影响模型的可转移性和泛化能力。此外,原子分解的计算成本仍与原子数线性相关,尚难进一步降低。
另一种策略是基于多体展开的机器学习势,其将体系总能量分解为一体、二体、三体等多体能量之和,并以此构建高精度模型,尤其在水体系模拟中表现优异。但随着高阶多体项的快速增长,该方法在大规模和长时间模拟中也面临计算瓶颈。
为解决上述问题,研究人员提出了一种融合两种方法优势的新型机器学习框架。该框架基于单体能量分解,将势能表示为具有化学意义的单体级别的一体和二体能量之和,避免了原子局部能量的不确定性。通过引入置换不变多项式(PIPs)作为神经网络的输入,确保结构描述符的对称性和物理合理性。研究人员将该方法命名为 MB-PIPNet,并在多个体系中进行了评估,包括气相簇、水-甲烷簇、液态水和液态二氧化碳。
结果表明,该方法仅凭一体和二体描述符即可有效刻画三体等高阶相互作用,大幅提升了模拟效率,并在液态水分子动力学模拟中展现出良好的性能和计算可扩展性。该框架具备良好的通用性,适用于更复杂的分子体系,并对相关挑战和潜在解决方案进行了讨论。
结果
单体神经网络模型(MB-PIPNet)
研究人员提出的 MB-PIPNet 框架以单体为基本单位,将整个分子体系划分为若干单体,并将体系总能量表示为各单体受环境扰动后的能量之和。每个单体的能量由前馈神经网络预测,其输入包括两个部分:一是反映单体内部结构的一体描述符,二是体现周围分子环境作用的二体描述符,均基于置换不变多项式(PIPs)构建,确保物理对称性。这种表示方式既保留了物理解释性,又在效率上显著优于传统基于原子的神经网络势能模型。
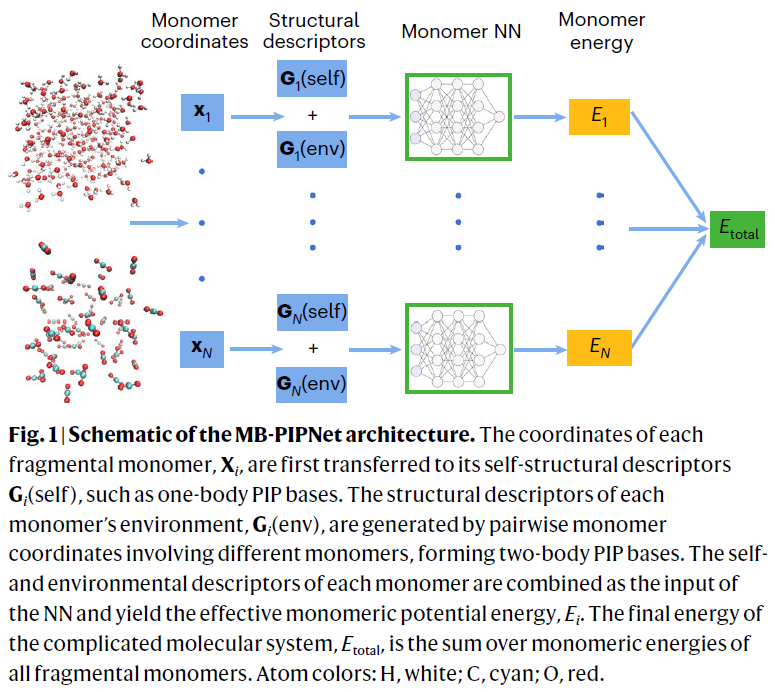
气相水三聚体的测试结果
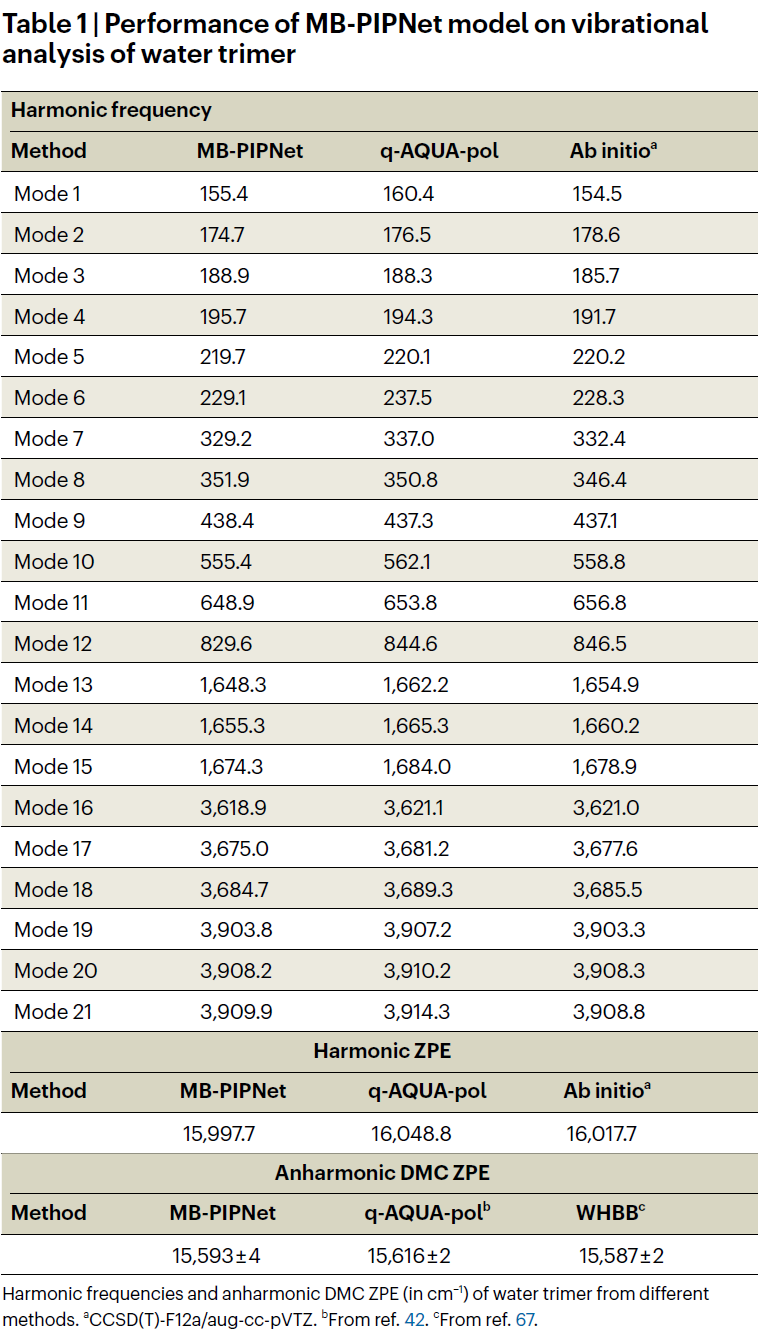
为验证该方法是否能有效捕捉多体相互作用,研究人员首先在水三聚体上进行了测试。基于高精度电子结构数据训练的模型,在能量预测中达到了极高的准确性。进一步通过谐振频率与量子力学模拟验证,该势能面不仅光滑且无数值“空洞”,表明该模型在描述复杂三体相互作用时表现出色。
液态水体系的扩展应用
研究人员进一步将 MB-PIPNet 应用于液态水体系,并使用多种量子化学方法计算的结构构建训练集。与现有的机器学习势能模型相比,MB-PIPNet 在能量和力的预测上均表现优越,尤其在无需复杂信息传递机制的前提下已具备较高精度。
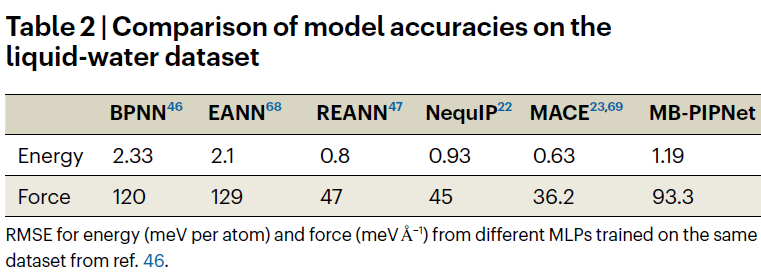
在更大规模数据集上训练后的模型,其能量预测误差低于现有主流模型(如 DeePMD),并可合理区分液态水中不同分子的结构扰动与相互作用所导致的单体能量差异,充分展现了模型在复杂体系中的泛化能力。
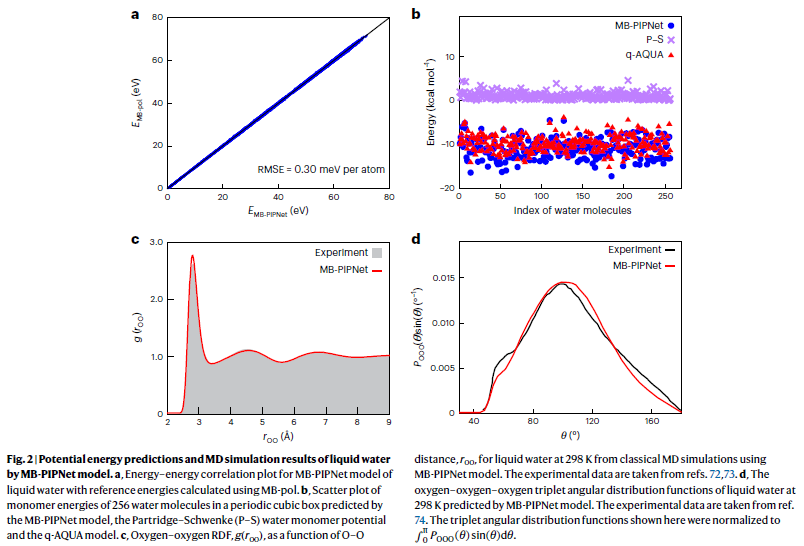
液态水的结构与动力学预测
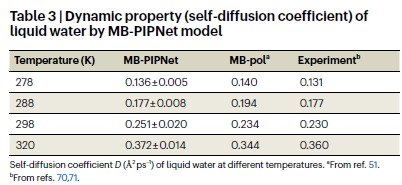
研究人员利用该模型进行液态水的分子动力学模拟,预测得到的径向分布函数在不同温度下均与实验数据保持高度一致,证明其对结构特性的良好再现能力。
此外,该模型还成功复现了液态水中典型的三体角分布和温度相关的自扩散系数,进一步证实其在描述液态体系静态与动态性质方面的准确性与可靠性。
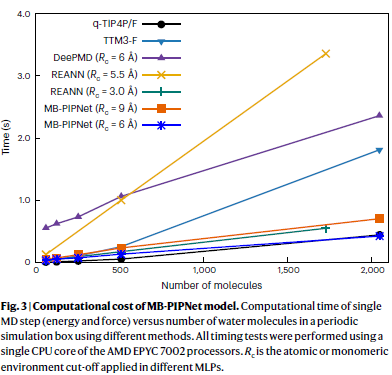
力场级的计算效率
前文已展示 MB-PIPNet 方法在从气相簇到凝聚态体系中对多体相互作用的准确刻画能力。其以化学启发的单体能量为核心构建方式,相较于传统原子分解势更具物理意义。此外,MB-PIPNet 还具备优异的计算扩展性与效率,主要源于两个方面:一是结构描述符采用了置换不变多项式(PIPs),已在多个体系中被验证为高效且系统性强;二是该方法将总能量表示为各单体能量之和,使得计算开销随着分子数量线性增长,而非原子数量。
在液态水模拟中,研究人员将 MB-PIPNet 的计算效率与多种传统力场(如 TTM3-F、q-TIP4P/F)以及主流 MLP 模型(如 DeePMD、REANN)进行了比较。在使用单个 CPU 核心进行能量与梯度计算的测试中,MB-PIPNet 显示出线性扩展性,且在一定截断距离设置下,其计算速度可与极化水力场 TTM3-F 相当,甚至超过 DeePMD 模型。
当模拟体系达到数千个水分子时,MB-PIPNet 不仅避免了代价高昂的静电求和操作,还在计算效率上超过 TTM3-F,接近非极化力场 q-TIP4P/F 的水平。在使用更短截断距离时,MB-PIPNet 的性能进一步提升,表现出接近甚至超过常用非极化力场的速度。
尽管某些先进的消息传递神经网络模型(如 MACE)在使用 GPU 并行计算时可达到与 MB-PIPNet 类似的性能,但值得注意的是,MB-PIPNet 在无需 GPU 的单核 CPU 环境下已具备这一效率,表明其在并行化或 GPU 加速下还有巨大提升潜力。通过合理调整截断范围,在保证精度的同时提升效率,这一策略同样适用于 MB-PIPNet 框架。
其他分子体系中的表现
在展示水三聚体与液态水的结果之后,研究人员进一步测试了 MB-PIPNet 在其他体系中的可迁移性。对于甲烷-水混合簇体系,模型在训练与测试集上均达到了较小的误差,表明其在描述异质气相体系方面同样有效,且计算成本依然呈线性扩展。
在液态二氧化碳体系中,仅使用 2,687 个基于 BLYP-D3 方法计算的构象数据进行训练,MB-PIPNet 模型就实现了较高的预测精度。这一结果进一步验证了该方法对凝聚态分子体系的适应性与灵活性。不过,为确保泛化能力,仍需结合更大规模的训练集与动力学性质评估进行后续研究。
值得一提的是,MB-PIPNet 的通用性还得益于背后强大的 PIP 库支持,该库已被用于开发超过 100 个不同分子的机器学习势能模型。除 PIPs 外,研究人员也可选择使用其简化版本——基本不变量(FIs),在高维体系中表现良好,且生成过程系统规范。已有研究表明,FIs 在处理原子数超过 20 或 30 的大体系时具有良好拓展性。
讨论
MB-PIPNet 框架摒弃了传统广泛使用的原子局部能量分解方法,转而将总势能表示为具有化学意义的单体能量之和。不同于标准的多体展开策略,MB-PIPNet 通过结合一体和二体的 PIP 描述符,有效引入来自其他单体的多体扰动效应,从而避免了高阶多体项的昂贵计算,大幅提升了模型评估的效率。
这一设计为在复杂体系(如分子材料)中以接近第一性原理精度、同时具备传统力场级速度的模拟提供了可能。迄今为止,哪怕是诸如 DeePMD 等先进原子级 MLP 方法,也尚未实现如此理想的精度与效率平衡。
在液态水等凝聚态体系的训练中,MB-PIPNet 引入了二体环境描述符的截断距离。不过,当前方法尚缺乏对远程效应的明确建模,与 DeePMD 等其他 MLP 方法存在相似局限。未来可尝试引入消息传递机制以改进结构描述符的构建。
此外,MB-PIPNet 在复杂体系中的单体划分仍需合理设计。目前该方法在气相簇、分子液体等体系中适用性较强,但若要扩展至反应体系、大分子、生物分子或固体材料等,仍需进一步开发与优化。
对大型有机分子或生物分子,MB-PIPNet 可通过以下方向提升适用性:(1)优化 PIP 或 FI 理论,使其支持 20 原子以上体系的多项式构建;(2)发展自动分割策略,将大分子拆解为更小、更易处理的子片段。
对于凝聚态反应体系或材料体系,如固态氧化物,采用原子级 MLP 往往更为自然,MB-PIPNet 的直接应用较为困难。一种可行的方向是将 MB-PIPNet 与原子级模型相结合:例如在 CO₂ 与水反应生成碳酸的模拟中,可用原子级模型刻画反应区,而用 MB-PIPNet 描述非反应的溶剂水分子,从而在保证精度的同时加快模拟效率。这类混合框架也适用于催化剂或材料-分子相互作用的研究。
MB-PIPNet 所提出的以单体为中心的思路,也有望与其他类型的机器学习模型(如不变神经网络、等变神经网络)相结合,扩展其在计算化学、物理、材料科学与生物体系中的应用。研究人员希望该方法能推动机器学习势能模型的发展,为复杂体系的经典与量子模拟提供具备从头计算精度和力场级速度的解决方案。
整理 | WJM
参考资料
Yu, Q., Ma, R., Qu, C. et al. Extending atomic decomposition and many-body representation with a chemistry-motivated approach to machine learning potentials. Nat Comput Sci (2025).
https://doi.org/10.1038/s43588-025-00790-0
内容中包含的图片若涉及版权问题,请及时与我们联系删除

评论
沙发等你来抢