
DRUGAI
今天为大家介绍的是来自西北工业大学的施建宇教授、尚洁副教授以及中国中医科学院中医临床基础医学研究所的吕诚研究员(副所长)所带领的团队发表的一篇论文。在新分子设计领域,如何推断合适的合成反应路径(即逆合成)是一个重要课题。近年来,计算方法在单步逆合成预测方面取得了可喜的进展。不过,现有方法都存在一些局限性:基于模板的方法受限于已知的合成模板库;无模板的方法难以解释其预测结果;而半模板的方法则未能充分利用化学实体之间的关联关系。为了解决这些问题,本文开发了一个创新的多任务图表示学习模型Retro-MTGR,用于单步逆合成预测。该模型充分利用了三种关联关系:合成子之间的内部关联、合成子与离去基团之间的相互关联以及离去基团之间的内部关联。这使得模型能够同时完成反应中心的推导和离去基团的识别这两项任务。
通过与16种最先进的方法进行对比,结果证明了Retro-MTGR模型的优越性。研究还验证了该模型的稳定性、可扩展性,以及各个关键组件的重要作用。更值得注意的是,该模型不仅能够判断某个化学键是否可以作为反应中心,还能为特定的合成子推荐合适的离去基团。这些发现反映了基础的化学合成规律,尤其是化学实体(如反应位点、合成子和离去基团)之间的电性互补关系。通过实际案例分析,研究表明Retro-MTGR推断的逆合成路径在单步合成反应中具有良好的应用前景。

现代药物设计领域,人工智能技术在靶点筛选、分子生成和药物性质预测等方面取得了显著进展,大大降低了研发成本和时间。其中,逆合成推断是一个关键环节,它能够帮助研究人员确定如何从现有原料合成目标分子,是将计算机模拟转化为实验室实践的重要桥梁。目前,逆合成预测主要有三种方法,分别是依赖已知的合成模板库的基于模板的方法、直接预测合成路径的无模板方法以及结合了前两种方法的优点的半模板的方法。
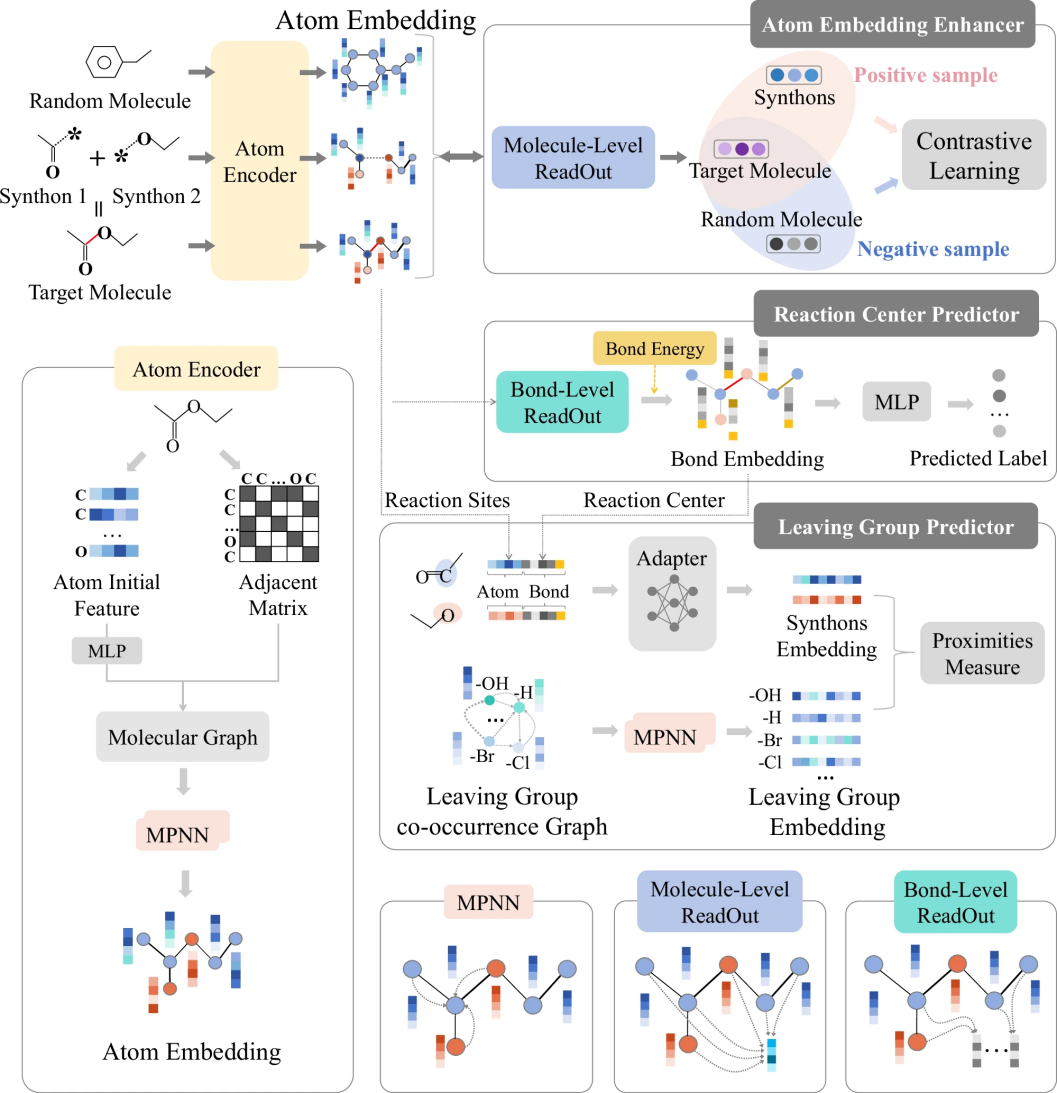
图 1
图1展示了本研究提出的创新性多任务图表示学习框架(Retro-MTGR)。该框架包含三个核心部分:
原子特征增强器(AEE):通过对比学习技术,分析目标分子与其组成部分(合成子)的结构异同
反应中心预测器(RCP):结合化学键能量等基础知识,准确识别可能发生反应的化学键位置
离去基团预测器(LGP):建立一个统一的特征空间,用于分析反应物组分(合成子)和离去基团的匹配关系
该框架特别重视化学反应中三种重要的关联关系:合成子之间如何组合形成目标分子、不同离去基团之间的相互作用以及合成子与离去基团之间的配对关系。这种设计充分考虑了实际化学反应中的规律,比如简单的离去基团(如H、Cl、Br、OH等)往往更容易参与反应,而能量较高的化学键则不容易作为反应中心。通过分析这些关联关系,该框架能够更准确地预测逆合成路径,并且预测结果具有更好的可解释性。
本研究的主要发现
通过分析化学键的特征分布(基于美国专利商标局的50K和480K化学反应数据集),研究发现:
能量较高(≥360 kJ/mol)的化学键,包括双键(如碳-碳双键、碳-氧双键、碳-氮双键)、三键(如碳-碳三键、碳-氮三键)和芳香键,通常不会参与反应,形成独立的特征类别
能量较低(<360 kJ/mol)的单键(如碳-碳单键、碳-氧单键等)可能会成为反应中心,这主要取决于分子的结构特征
通过分析化学键中原子对的电性(即得失电子能力)分布发现,当键中的两个原子具有相反的电性时,该键更可能成为反应中心
通过分析反应物各组分在特征空间中的分布规律,研究发现:
参与同一反应的两个合成子(反应物的主体部分)具有相反的电性,且在特征空间中距离较远
在同一反应物中,合成子和离去基团具有相反的电性,且在特征空间中距离较近
同一反应中的两个离去基团通常具有相反的电性,且在特征空间中距离较远
常见的离去基团(如氢、氯、溴、碘和羟基)通常结构简单,在特征空间中分布分散;而特定反应专用的离去基团则会按照反应类型聚集在一起,它们之间的结构也较为相似。
数据集与参数设置
研究使用了来自美国专利商标局(USPTO)的化学反应数据集,包含了50,016个详细记录了原子变化过程的化学反应(称为USPTO-50K数据集)。这些反应可以分为两大类:广义偶联反应(两个分子结合形成新分子)和其他类型反应(如保护、还原等)。研究发现,在复杂的多步合成过程中,广义偶联反应占据了超过80%的比例,因此本研究重点关注这类反应。
在筛选后,研究最终使用了35,682个广义偶联反应数据,这些数据被分为三部分:80%用于训练模型、10%用于验证模型,最后10%用于测试模型的性能。
在模型参数设置方面,Retro-MTGR使用28个不同维度来描述原子的特性,包括:
原子类型(23个特征)
氢原子数量(1个特征)
相邻原子数量(1个特征)
是否为芳香性(1个特征)
电荷(1个特征)
原子质量(1个特征)
为了提高模型性能,研究采用了多层神经网络来处理这些特征。通过实验发现,使用两层神经网络能够达到最佳效果,这一结论在已知和未知反应类型的情况下都成立。
在预测反应中心时,模型考虑了33个特征,包括32个结构特征和1个化学键能量特征。对于离去基团的预测,模型根据不同反应类型采用了相应的特征空间。最后,通过反复实验确定了最优的任务权重配比:主任务占60%,两个辅助任务各占20%。
实验结果
研究团队将新开发的 Retro-MTGR 系统与16种最先进的化学反应预测方法进行了比较(4种基于模板的方法、6种无模板的方法以及6种基于半模板的方法)。
表1:与SOTA方法在Top-k精度上进行比较
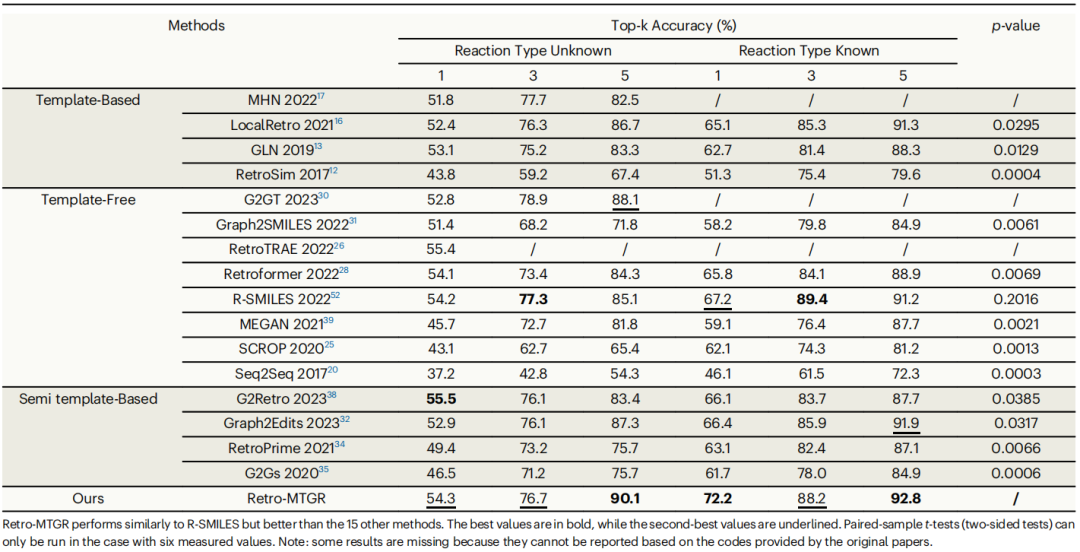
实验分两种情况进行:一种是完全不知道反应类型的“盲测”(RTU),另一种是已知可能发生的反应类型(RTK)。结果表明,在没有反应类型提示的情况下,Retro-MTGR 能以54.3%的准确率预测出正确的反应路径,如果给出3次预测机会,准确率可提升至76.7%,给出5次机会则可达到90.1%。对于已知反应类型的情况下,这些数字分别提升到72.2%、88.2%和92.8%。
为了进一步验证系统的实用性,研究人员还在更大规模的USPTO-480K化学反应数据库上进行了测试。这个数据库包含了近50万个真实的化学反应案例。在这个更具挑战性的测试中,Retro-MTGR 依然表现出色,其预测准确率显著超过了目前最先进的R-SMILES模型。统计分析表明,Retro-MTGR在各项指标上的优势具有统计学意义。
消融实验
研究团队通过消融实验深入分析了Retro-MTGR系统的各个核心部分。他们创建了7个简化版本的系统,每个版本都移除了一个关键功能,就像解剖手术一样逐一研究每个组件的作用。这些简化版本包括:去除多任务协同学习功能、移除原子特征增强模块、不考虑化学键的能量和类型信息、移除离去基团(化学反应中会分离出去的原子团)关联分析、以及删除一些技术优化手段。
表2:消融对比
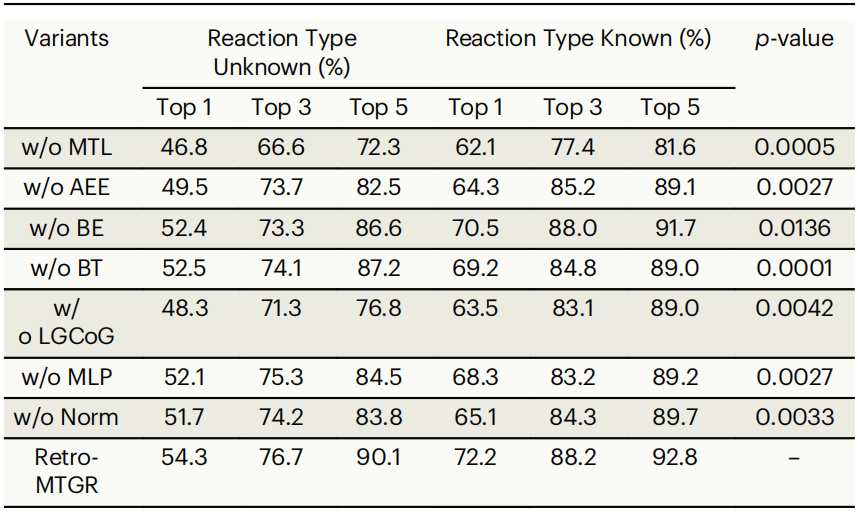
表2显示,完整的Retro-MTGR系统性能显著优于所有简化版本。其中最关键的是多任务协同学习功能:当系统需要在完全不知道反应类型的情况下进行预测时,有了这个功能可以将准确率提高7.5%到17.8%不等。这说明让系统同时学习“在哪里切断分子”和“会分离出什么基团”这两个任务,能帮助它更好地理解化学反应规律。
第二重要的是离去基团关联分析功能,它让系统能够学习到哪些原子团倾向于一起参与反应,这个功能为准确率带来了3.8%到13.3%的提升。原子特征增强模块排在第三位,通过更好地理解原子的性质,使准确率提高了3.0%到7.9%。研究还发现,即使是看似简单的化学键能量信息也能显著提升系统性能,因为高能量的化学键往往更容易参与反应。此外,一些技术层面的优化手段,如使用更丰富的原子表示方法和数据标准化处理,也都对提升系统性能起到了重要作用。
化学规则的发现
研究团队深入分析了Retro-MTGR系统是如何“学会”化学反应规律的。他们以常见的酯化反应为例,揭示了一个有趣的发现:化学反应往往发生在带有相反电性的原子之间。就像磁铁的南北极会相互吸引一样,一个倾向于“索取”电子的原子(带正电性)会与一个倾向于“给出”电子的原子(带负电性)发生反应。
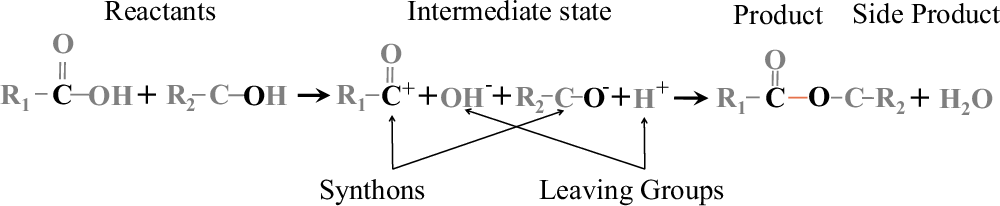
图 2
图2形象地展示了这种“相反相吸”的规律。研究发现,在化学反应中存在三种这样的配对:形成主产物的原子对、形成副产物的原子对,以及参与反应的原子与其分离基团之间的配对。这些配对都遵循着相同的电性互补原则。
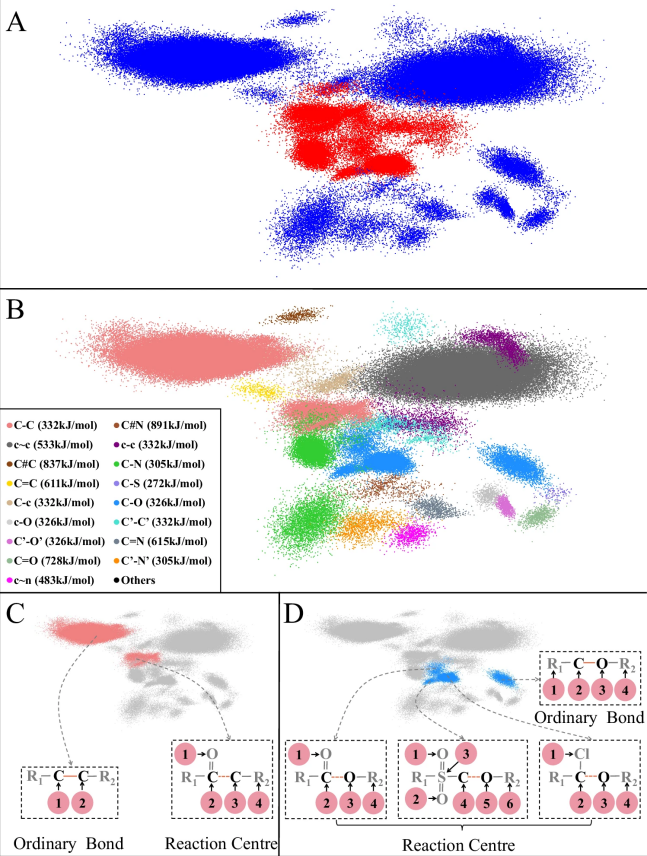
图 3
为了更深入地理解这一规律,研究人员分析了化学键的特征。如图3所示,他们发现:
高能量的化学键(如三重键、芳香键)通常很稳定,不易参与反应。
低能量的单键可能会发生反应,但具体要看周围原子的影响。
模型能够自动学习到这些规律,并在预测时准确区分可能发生反应的位置。
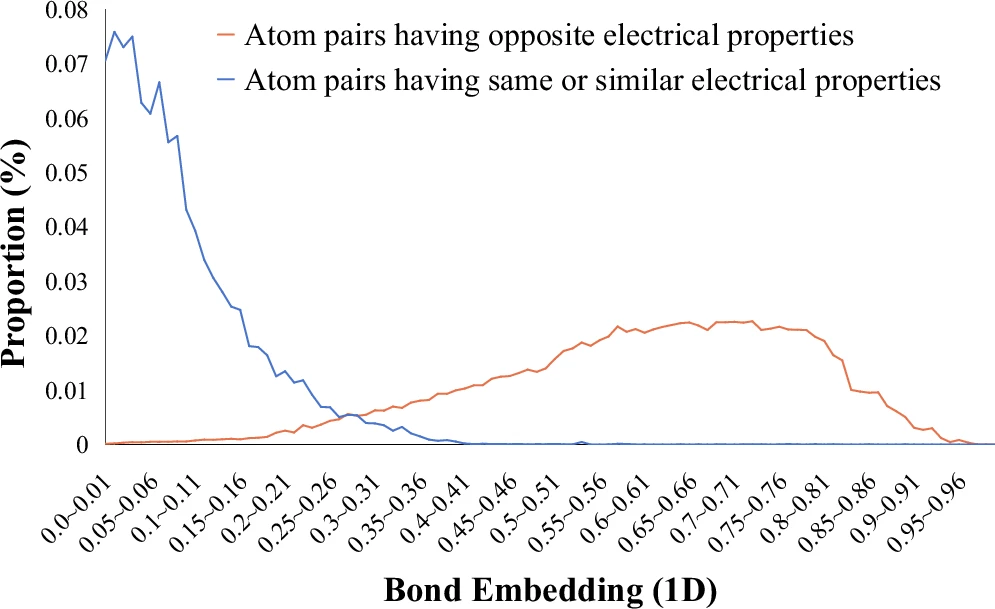
图 4
更有趣的是,研究发现同样类型的化学键在不同分子中可能表现出不同的反应倾向。这就像一个人的性格不仅取决于自身,还受周围环境的影响。图4证实了这一点:当一个化学键连接的两个原子具有相反的电性时,它更可能成为反应发生的位置。这些发现不仅证明了Retro-MTGR模型确实“理解”了化学反应的基本规律,而且这种理解方式与化学家们长期积累的经验知识高度一致。这意味着AI系统不仅能预测化学反应,还能帮助我们更好地理解化学反应的本质。
合成子与离去基团的联合视图
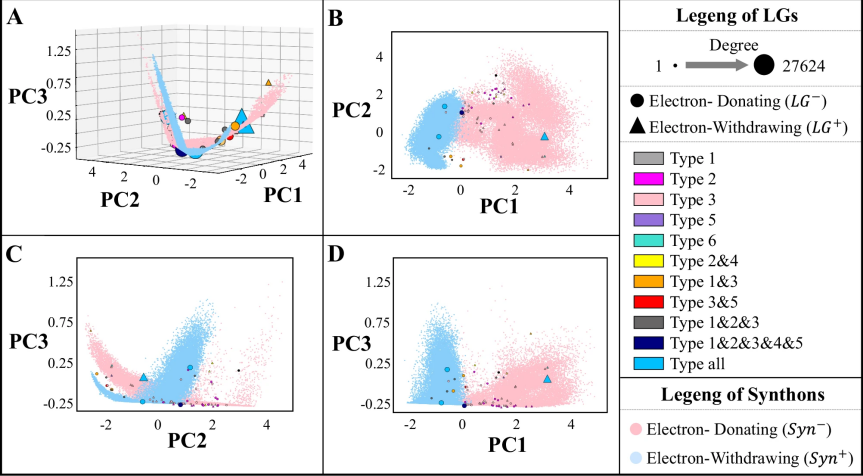
图 5
研究团队通过分析Retro-MTGR系统的“思维空间”,揭示了化学反应中各个参与者之间的关系规律。为了便于理解,他们使用了一种叫做PCA的数学工具,将复杂的高维数据转换成可以在3D空间中展示的图像(图5)。
这个分析揭示了四个重要发现:
首先,在化学反应中,两个反应物(合成子)之间存在明显的“相异相吸”规律。就像磁铁的南北极一样,带正电性的合成子(蓝点)和带负电性的合成子(红点)会相互吸引。数据显示,超过94%的合成子都遵循这一规律。图6A显示,参与同一反应的合成子在空间中的距离明显大于不参与同一反应的合成子,这进一步证实了它们的互补性。
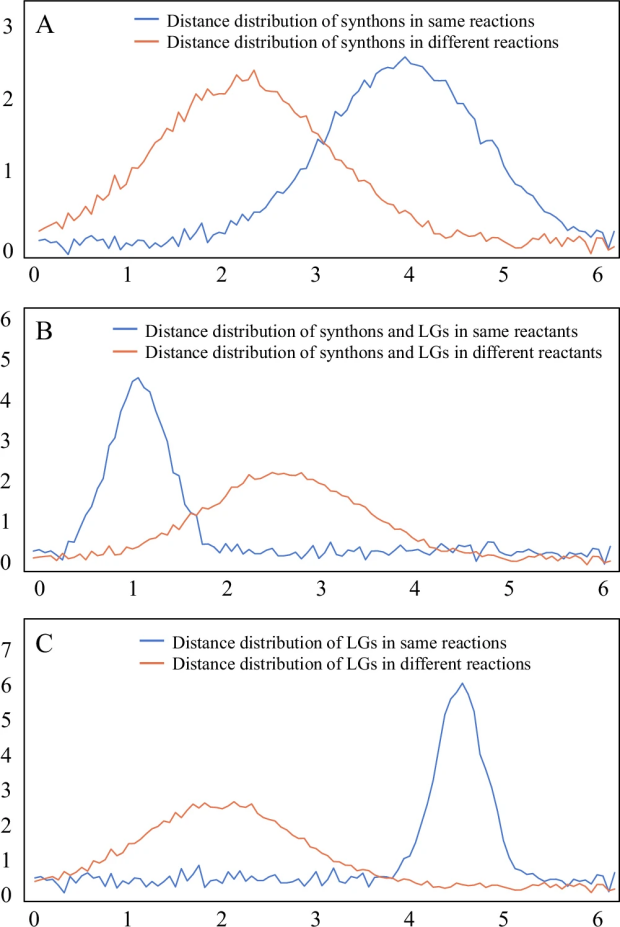
图 6
其次,研究发现反应物和它所释放的基团之间也存在规律:它们往往带有相反的电性,就像拼图的两个部分一样相互匹配。约77%的情况都符合这一特征。图6B展示了这种关系:同一反应物中的合成子与离去基团距离明显小于来自不同反应物的配对。
第三,关于离去基团的规律特别有趣。研究发现它们分为两类:
“常见基团”:如氢(H)、羟基(OH)、氯(Cl)等简单结构,它们就像化学反应中的“万能配角”,可以参与多种类型的反应;
“专业基团”:结构较复杂,只在特定类型的反应中出现,就像是“专业演员”只在特定场景出现。
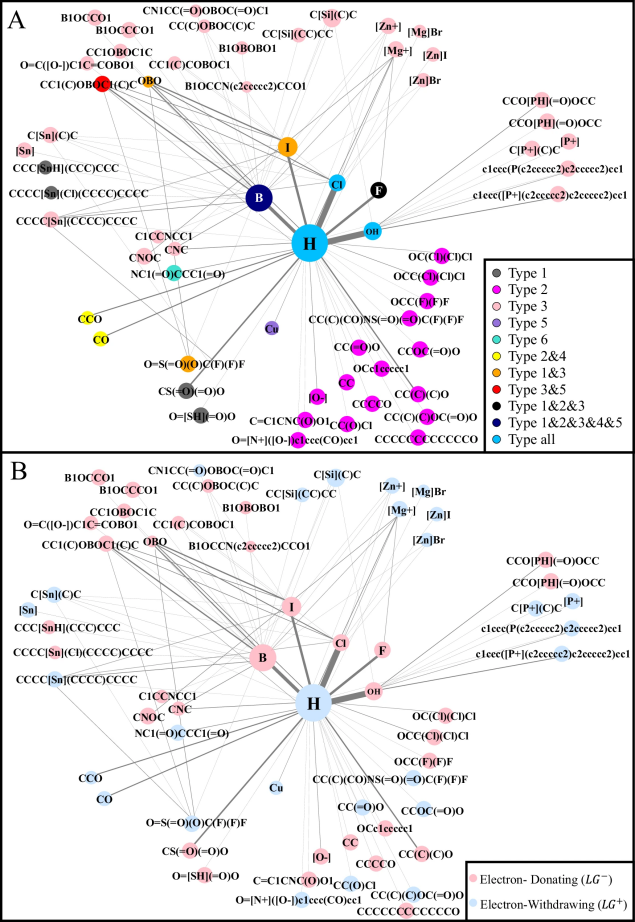
图 7
图7A清晰地展示了不同类型反应中离去基团的分布情况,而图7B则展示了它们的电性分布。
最后,两个离去基团之间也存在明显的规律:超过94%的配对具有相反的电性。最常见的组合是氢与羟基(占27.8%)、氢与氯(占27.4%)等简单基团的配对。图6C显示,经常一起出现的离去基团对在空间中的距离明显大于不常一起出现的配对。
案例研究
研究团队选择了两种市面上的新药来测试他们的AI系统:一种是用于治疗皮肤癌的Sonidegib,另一种是用于治疗消化不适的Acotiamide。这两种药都不在系统的训练数据中,因此非常适合用来验证系统的实际应用能力。
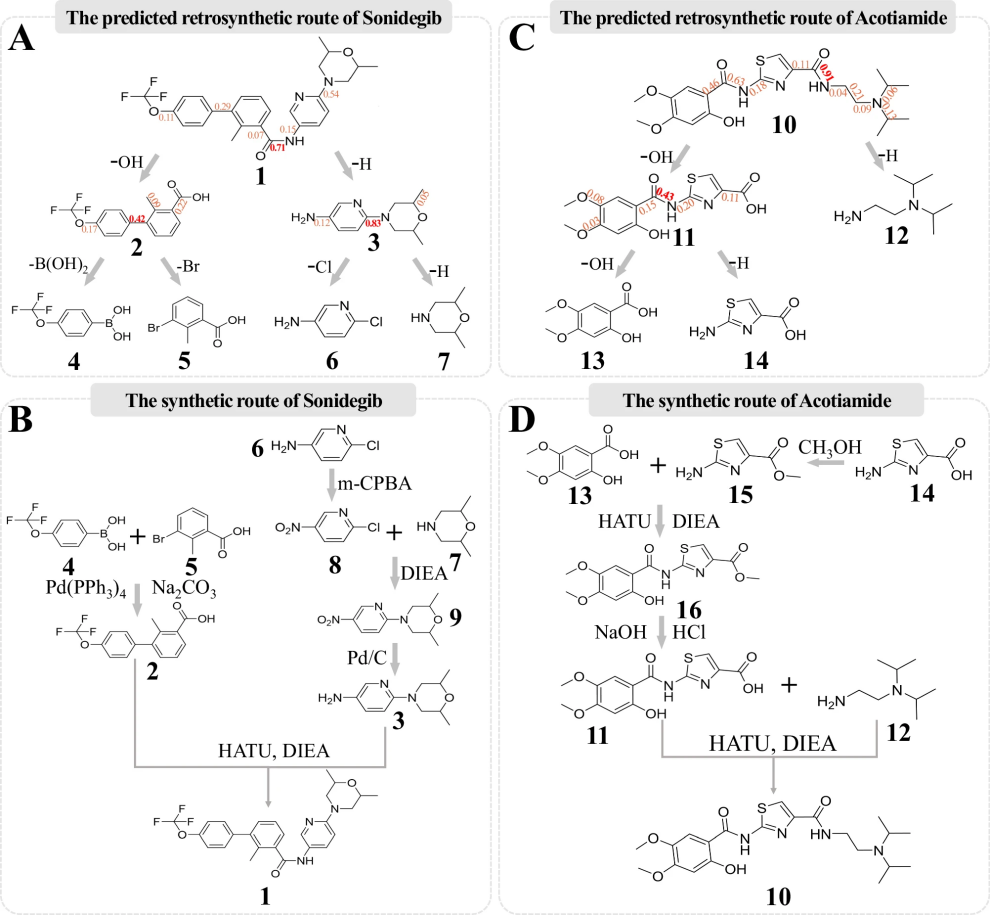
图 8
图8生动地展示了AI系统是如何“解构”这两种药物分子的:
对于Sonidegib(图8A),系统就像在玩乐高积木一样,准确地找到了将大分子拆分成更小部件的方法。它先将目标分子(标记为“1”)分成两块(“2”和“3”),然后继续将这些部分分解成更小的片段(“4”到“7”)。这就像提供了一份“倒序”的组装说明书。当化学家们按照AI的建议进行实际合成实验时(图8B),发现了一个有趣的问题:某些分子片段(“6”)会自己与自己反应,这就像有些乐高积木会不小心自己粘在一起。科学家们巧妙地解决了这个问题:他们先将这些“自我粘连”的片段做了一点改装(变为“8”),等完成需要的反应后,再将它们变回原来的样子。
对于Acotiamide(图8C和D),系统同样展示出了出色的预测能力。它成功预测出了将大分子(“10”)分解成更小片段(“11”到“14”)的路径。在实际合成过程中,科学家们也遇到了类似的挑战,并通过将某些分子暂时改装成不同的形式(如将“14”变成“15”)来解决问题。
这些案例研究证明,AI系统不仅能够准确预测化学反应路径,而且其预测结果在实际实验中是可行的。虽然有时候需要科学家们做一些创造性的调整,但系统提供的基本路线是正确的。这就像GPS导航系统,虽然有时候需要根据实际路况做一些调整,但总体方向是准确的。这项研究为未来的药物合成提供了一个强大的规划工具。
讨论
本文介绍了一个创新的AI化学合成预测系统Retro-MTGR。研究表明,该系统在与其他16种先进方法的对比中展现出显著优势,不仅能准确预测化学反应的关键步骤,还能解释为什么某些化学键会成为反应中心,以及哪些离去基团最适合特定的合成子。系统的核心创新在于结合了多任务学习框架、离去基团共现图和基于图对比学习的原子嵌入增强等技术,使其能够全面理解和预测化学反应规律。
虽然系统在两个新药物案例中展示了良好的实际应用价值,但研究团队也坦承系统存在一些待改进的问题:离去基团在训练数据中分布不均衡、多步合成预测能力有限、缺乏真实键能数据等。这些局限性为未来的研究指明了方向,特别是在整合更多合成因素(如反应产率、条件和试剂)方面,有望发展成为更完善的多步逆合成路径规划工具。
编译|于洲
审稿|王梓旭
参考资料
Zhao P C, Wei X X, Wang Q, et al. Single-step retrosynthesis prediction via multitask graph representation learning[J]. Nature Communications, 2025, 16(1): 814.
代码:
https://doi.org/10.5281/zenodo.14346324
内容中包含的图片若涉及版权问题,请及时与我们联系删除

评论
沙发等你来抢