
DRUGAI
疾病发生和进展的特异性研究中,识别调控转录因子(TRs)仍具有挑战性,这些因子通过调控元件和表观基因组信号控制基因表达。大规模多组学表观基因组数据的引入,为解析调控元件及其调控因子的复杂模式提供了可能。研究人员在此提出TRAPT,一个多模态深度学习框架,可通过学习和整合靶基因的顺式调控元件及全基因组结合位点的调控潜能,推断转录因子的活性。在570个与TR相关的数据集上,TRAPT在预测转录因子,尤其是协同因子和染色质调控因子方面优于现有方法。此外,该方法成功识别出与疾病、遗传变异、细胞命运决策及组织相关的关键转录因子,展现了基于表观组数据识别TRs的全新视角。

基因表达调控由多种上游转录调控因子(TRs)共同驱动,包括转录因子(TFs)、协同因子(TcoFs)和染色质调控因子(CRs),它们介导启动子与远端增强子的信号传递。异常的基因表达常与疾病发生密切相关,因此识别关键调控因子对于理解疾病机制至关重要。随着 ChIP-seq 和 ATAC-seq 等技术的发展,顺式与反式调控图谱得以清晰描绘。TRs 在染色质开放性和组蛋白修饰等表观信息的共同作用下,展现出细胞特异性的调控活性。此外,大量研究表明 TFs 可通过结合启动子或增强子等顺式调控序列,调节靶基因表达。鉴于基因调控机制的复杂性,研究人员亟需利用大规模表观基因组数据,全面捕捉基因上游的协同调控特征,以提升 TRs 的预测能力。
高通量测序技术的快速发展使 ATAC-seq、DNase-seq 和 ChIP-seq 等数据迅速积累。然而,不同来源的数据存在噪声干扰、批次效应及冗余等问题,增加了整合与表示学习的难度。目前已有多种方法尝试通过功能基因集推断上游 TRs,如 Enrichr、ChEA3、Lisa 等,但这些方法大多基于统计富集分析,未能充分利用顺式调控元件(CREs)的详细信息。而CREs是 TF 功能的核心,缺乏这部分信息将限制调控因子的准确预测。一些方法虽引入 CRE 数据,但仅局限于与输入基因集相关的区域,无法覆盖全基因组调控图谱。此外,TRs 往往倾向于结合在活跃染色质区域,表现出结合偏好(TRBP),这类偏好尚未被现有方法充分建模。更重要的是,当前大多数方法仅限于通过基因集推断上游调控因子,缺乏对其全基因组结合位点的推理能力,因此急需能够全面整合CRE与结合位点的预测框架。
多组学表观数据的整合也存在诸多挑战,例如跨模态干扰与非线性关系。现有方法多基于传统回归模型,未能有效处理跨模态间的复杂关系。深度学习技术在多个生物学任务中已展现出强大表现,其关键在于高质量数据的采集与建模。研究人员此前构建了多个表观调控数据库,如 TcoFBase、CRdb、TFTG、SEdb 和 ATACdb,系统汇聚了大量 TRs、增强子与染色质开放性信息,构建了当前最全面的表观特征资源库。结合这些数据资源与先进深度学习方法,为深入解析表观调控网络提供了前所未有的可能。
本研究中,研究人员提出了一个数据驱动的深度学习框架——TRAPT,融合多阶段学习结构,通过知识蒸馏和图卷积神经网络整合表观特征,联合建模靶基因的顺式调控元件信号与全基因组结合位点,优化 TR 活性表示,并识别上下文特异的关键调控因子。在570个 TR 敲除数据集上进行评估,TRAPT 在预测 TFs、TcoFs 和 CRs 方面均优于现有方法。进一步,TRAPT 成功识别出阿尔茨海默病中的关键因子(如 REST),并在细胞发育与组织数据中准确预测了控制细胞命运与组织特异性的调控因子。
结果
TRAPT 方法概述
TRAPT 是一个整合多组学信息的预测框架,旨在根据输入的感兴趣基因集推断转录调控因子(TRs)的活性。简而言之,该模型以基因集为输入,输出每个TR的活性评分。TRAPT 采用多阶段融合策略,分别处理来自全基因组结合位点的下游调控潜能(D-RP)以及源自顺式调控元件的上游调控潜能(U-RP),从而解决TR结合偏好(TRBP)和顺式调控信息缺失(ICCP)的问题。
TRAPT 的流程主要包括四个步骤:
计算表观调控潜能(Epi-RP)与转录因子调控潜能(TR-RP):研究人员收集了超过 20,000 份表观基因组数据,包括 ATAC-seq、H3K27ac 和 TR ChIP-seq 数据,并进行预处理后,计算每个基因的调控潜能。通过对表观组数据统一加权衰减,并对TRs采用上下文特异的衰减策略,分别得到 Epi-RP 和 TR-RP,用于后续建模。
预测 TR 的下游调控潜能(D-RP):该步骤旨在将 Epi-RP 与 TR-RP 融合,构建 TR 与表观样本之间的异构网络(ERN)。研究人员采用 k 近邻算法建立网络,并使用条件变分自编码器(CVAE)与图变分自编码器(VGAE)构建的知识蒸馏模型(D-RP模型)进行优化。教师模型学习多模态嵌入,学生模型学习统一、低噪声表示。该机制提升了模型对跨模态数据的鲁棒性,输出为 D-RP 矩阵。
预测查询基因集的上游调控潜能(U-RP):研究人员设计了另一个知识蒸馏模型(U-RP模型),以Epi-RP和输入基因集为基础,选出关键的表观组样本并推断其调控作用。学生模型在教师模型提供的“软标签”指导下,通过稀疏组套索(SGL)方法引入组内与组间稀疏性,实现对非冗余、非线性组合样本的精准筛选。输出为U-RP向量,包含与查询基因相关的染色质开放性(ATAC)与活性状态(H3K27ac)信息。
整合 D-RP 和 U-RP 推断 TR 活性:将归一化后的 TR-RP 和 D-RP 矩阵逐元素相加,再与 U-RP 向量逐元素相乘,获得整合调控潜能(I-RP)。随后,使用 ROC 曲线下面积(AUC)衡量 TR 与基因集的相关性,并合并两种路径得到最终的 TR 活性评分。
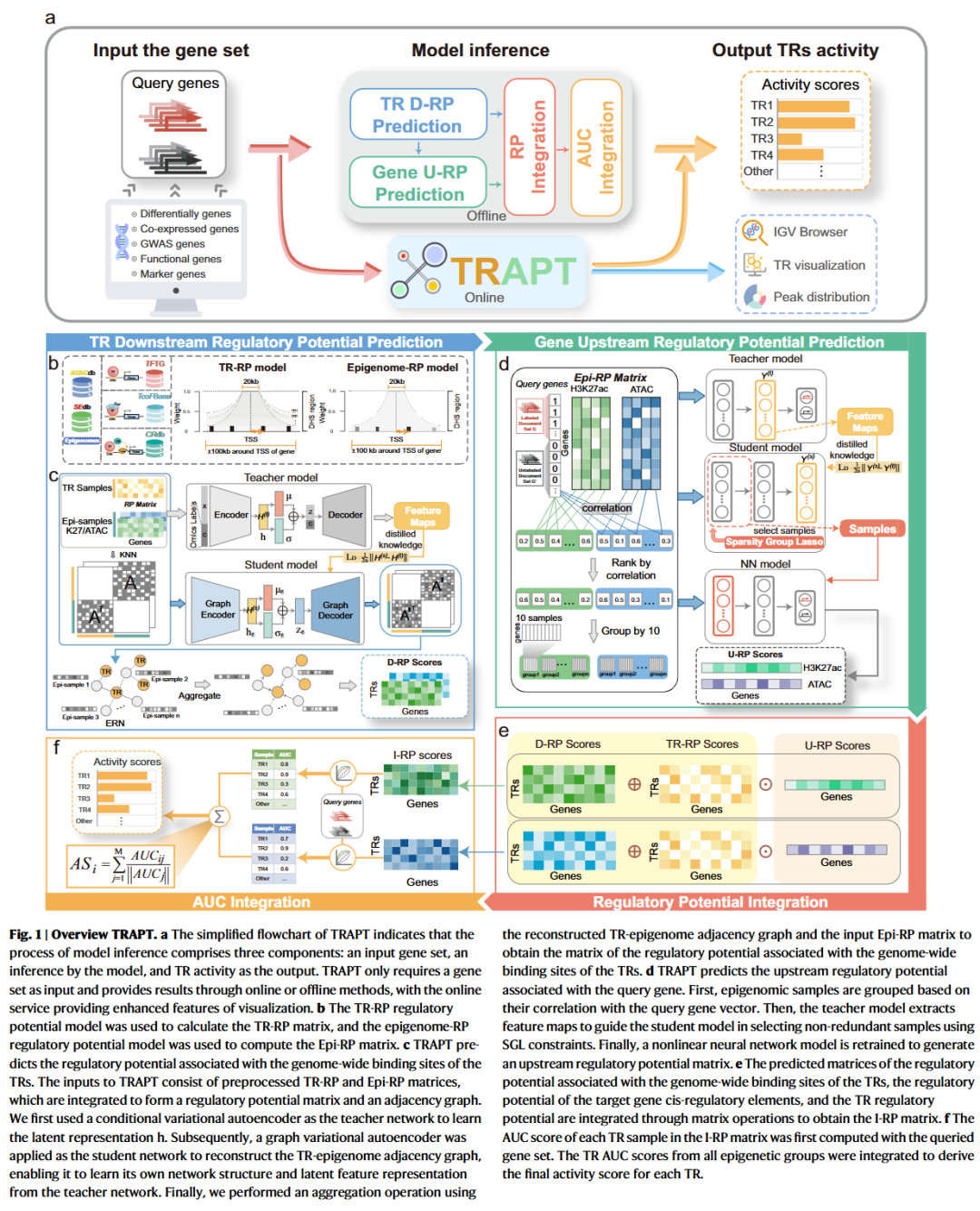
综上,TRAPT 融合了 TR 的全基因组结合潜能与靶基因顺式调控潜能,能够在上下文特异的基础上,高效识别调控输入基因集的关键 TRs。
TRAPT 在基准数据集上表现优越
研究人员采用“目标TR排序分数”作为主要评估指标,衡量算法根据基因集预测对应调控因子排名的能力。例如,在 GATA6 敲除实验中,若GATA6本身的排名靠前,则说明预测性能较好。为全面评估TRAPT,研究人员整合了来自 KnockTF 数据库的570个转录因子敲除/敲低数据集,经过质量控制和差异表达分析,从每个RNA-seq数据集中筛选出上调与下调基因,作为TRAPT的输入,并对预测结果中的目标TR排名进行评估。
研究人员将 TRAPT 与多个主流方法进行了比较,包括以基因集为输入的 Lisa、BART、i-cisTarget 及富集分析方法 ChEA3。评估标准包括命中前10或前N个目标TR的数量,以及整体TR识别表现。结果显示,TRAPT 在识别前10个TR方面比表现第二好的方法(Lisa)高出13%;相较 i-cisTarget 的提升更超过200%。TRAPT 的 AUC 达到 0.643,明显优于其他方法;其平均倒数排名(MRR)为 0.067,也分别比 Lisa 和 BART 提高了18% 和 76%。这些结果表明 TRAPT 在预测 TR 活性方面具备显著优势。
不同于以往方法主要集中在 TF 的预测,TRAPT 引入高质量的转录协同因子(TcoFs)与染色质调控因子(CRs)数据,使得其具备更全面的调控因子预测能力。在 TF、TcoF 与 CR 三类子集中,TRAPT 均展现出领先性能。特别是在 TcoF 与 CR 的预测中,Lisa 的表现显著下降,而 TRAPT 仍保持优异结果,说明其在多类调控因子预测上的全面性得益于多阶段融合策略与丰富的数据支持。
为了验证 TRAPT 的优势并非仅因数据覆盖度更高,研究人员将 Lisa 和 BART 与 TRAPT 一同使用 Cistrome 数据库的背景数据,并以 KnockTF 数据集为标准重新评估。结果显示,即便在相同背景数据下,TRAPT 的整体表现仍优于 Lisa(提升 30%)和 BART(提升 85.7%)。进一步评估显示,TRAPT 在 TF、CR 与 TcoF 三个类别中的AUC均高于对比方法,其中对 CR 的预测性能甚至比 Lisa 高出 198.4%。
此外,研究人员还比较了不同蛋白家族在不同类型数据(如 TR 敲除数据 vs. TR 结合数据)下的预测表现,发现部分家族(如 CP2、RXR-like)在敲除数据上表现更佳,而其他家族(如 zf-C2H2、IRF、THR-like)在结合数据中效果更好。这可能源于转录因子敲除后引发的二级转录调控效应。
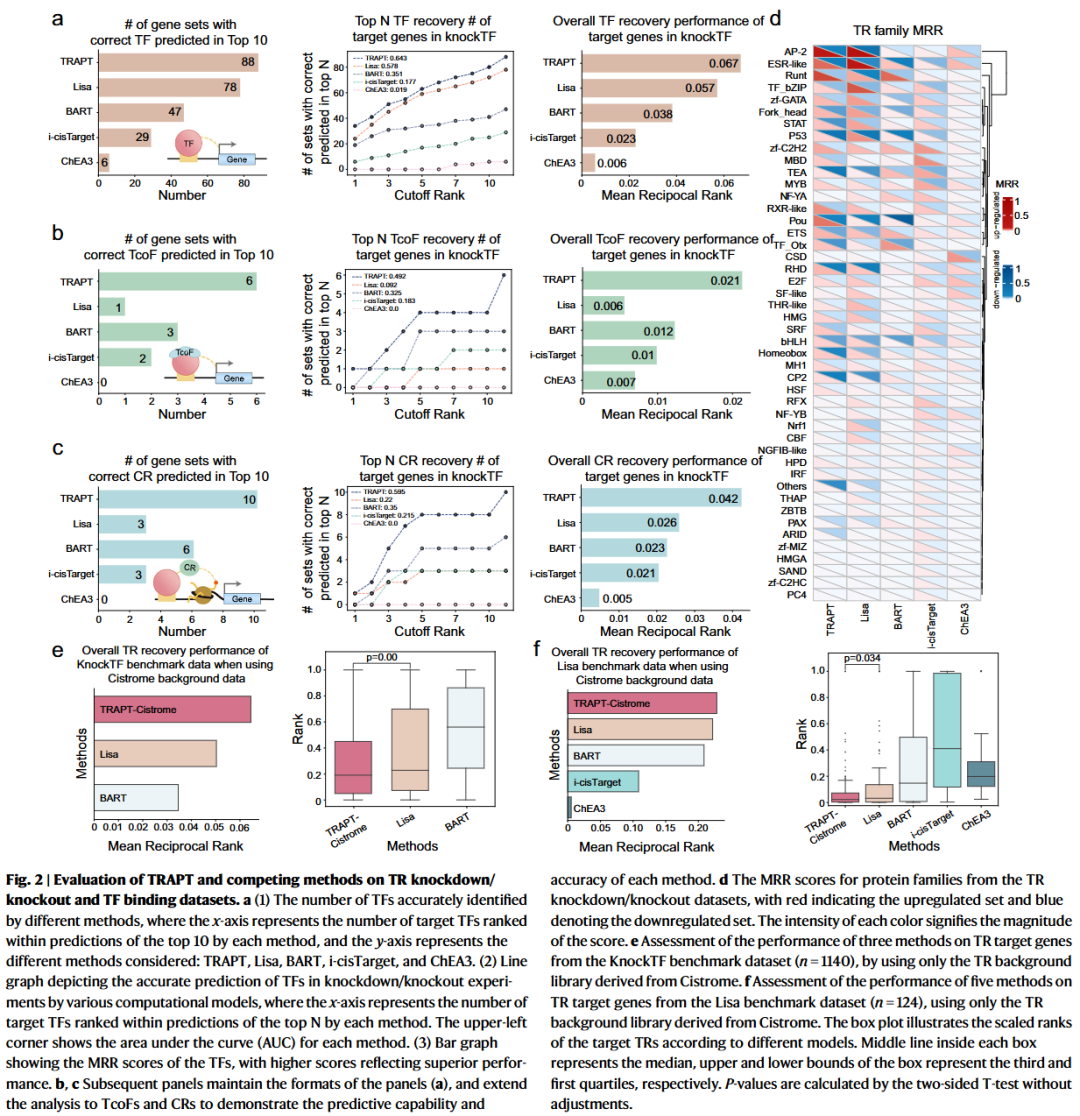
最后,针对大规模数据整合可能带来的运算时间问题,研究人员比较了 TRAPT 与 Lisa、BART 的运行时间。结果显示,TRAPT 不仅性能优越,还具有更快的运行效率,尤其在单个TR活性预测任务中显著快于其他方法。
多阶段融合策略显著提升转录调控因子预测性能
为验证多阶段融合策略对 TR 预测的提升效果,研究人员进行了系统的消融实验。TRAPT 中的 U-RP 模型用于模拟靶基因顺式调控元件的调控潜能,捕捉其上下文特异性的表观状态。当移除该模块时,整体性能显著下降,说明其有效表征了输入基因的表观调控特征。D-RP 模型则预测TR在不同条件下对基因组的结合偏好,考虑了调控元件的活性。去除该模块同样导致性能下降,进一步表明结合元件活性对于TR功能建模具有重要意义。此外,研究人员还通过计算TR与远端增强子结合比例,构建了特异性调控潜能模型。去除该TR特异性建模也导致性能下降,显示其对识别不同调控模式具有重要作用。
TRAPT 融合多种表观组学特征以预测最终的TR活性。对不同模块性能的深入分析发现,TRAPT-H3K27ac 模型对上调基因集预测更优,而TRAPT-ATAC 模型则更适合下调基因集。当移除所有表观模块,仅保留启动子区域模型时,预测性能显著下降。进一步逐步移除表观模块,整体性能也持续下降,表明TRAPT在多模态特征融合方面具有良好稳定性。
知识蒸馏机制在TRAPT中也发挥了重要作用。相比未使用知识蒸馏的模型(NKD),使用蒸馏策略(KD)在 KnockTF 数据集上提升了5.4%,在 Lisa 数据集上提升了7.9%。在top-10 TR预测任务中,KD模型的AUC高出8个百分点,表现显著优于NKD。模型在训练与验证集上的损失值快速下降,且蒸馏策略下 D-RP 学生模型收敛更快、准确率更高,表明知识蒸馏提升了模型稳定性且未引入过拟合。
在 D-RP 模型中,研究人员利用 kNN 算法构建TR与表观组学样本之间的连接网络,并以连接恢复任务评估其预测能力。实验结果显示,在验证集中,教师与学生网络的损失值迅速下降,最终在测试集上分别达到 auROC 0.81 与 auPRC 0.84 的表现。即便在最多屏蔽15%边的情况下,模型恢复能力依然稳定,AP 值保持在0.8以上,表明其对缺失连接具有良好的鲁棒性。
U-RP 模型的目标是基于基因集与 Epi-RP 矩阵预测顺式调控图谱。训练过程中,其损失值也稳定下降。为解决冗余样本筛选问题,研究人员分析了在不同数量样本下的性能变化。当特征数达到10时,性能提升趋于平稳,与已有研究一致。
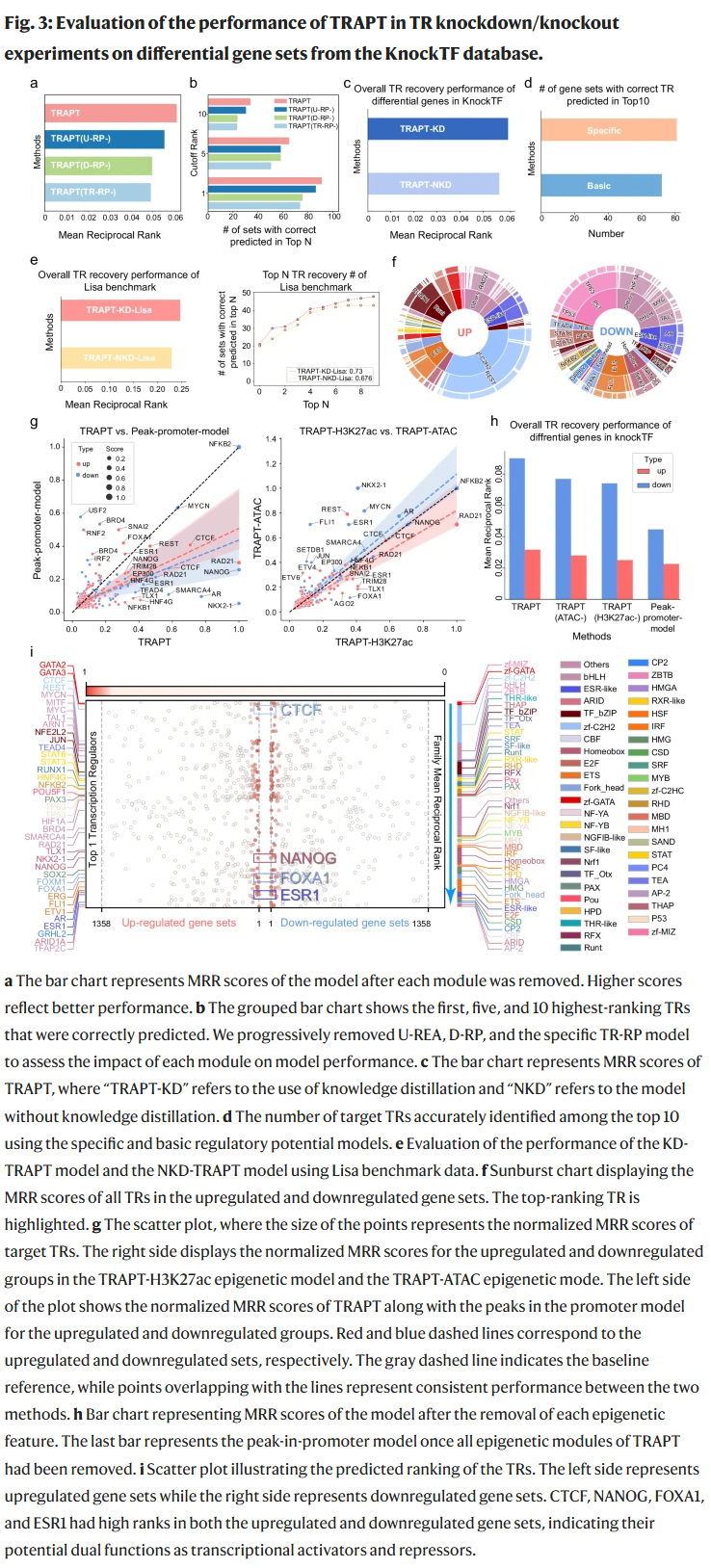
最后,研究人员比较了TRAPT在预测上调与下调基因集时的表现,发现其对下调基因集的预测更为准确。这一现象也支持了激活因子多于抑制因子的生物学观察。多数 TR 仅具有单一功能,部分如 CTCF、NANOG、FOXA1、ESR1 则同时具有激活与抑制作用。综上,TRAPT 能够准确识别激活型、抑制型及双功能调控因子。
TRAPT 在 ESR1 敲低实验中准确识别关键转录调控因子
ESR1 是乳腺癌 ER 阳性亚型中的关键转录因子,调控多种与疾病风险相关的下游基因表达。为验证 TRAPT 在疾病背景下识别关键 TRs 的能力,研究人员将其应用于 MCF7 ER+ 乳腺癌细胞中 siRNA 敲低 ESR1 后得到的差异表达基因集。TRAPT 准确预测到 ESR1 在下调基因集中排名第1,上调基因集中排名第17,反映出其在乳腺癌中兼具激活与抑制功能。
TRAPT 还识别出与 ESR1 密切相关的多个转录因子、协同因子与染色质调控因子,包括 FOXA1、EP300 和 MED1。其中,GATA3 是哺乳腺细胞命运的重要调控因子,FOXA1 通过调节染色质可及性影响乳腺癌发生发展,EP300 可乙酰化 ESR1,增强其靶基因表达。此外,从 STRING 数据库得到的这些高排名 TRs 之间具有频繁的相互作用;TCGA 和 GTEx 数据的共表达分析也显示 GATA3、FOXA1 和 ESR1 三者之间具有显著相关性,进一步验证了 TRAPT 的准确性。
研究人员还分析了 D-RP 分数对 TR 活性的刻画能力。将 ESR1 及其相关 TR 的 D-RP 分数分别作用于目标基因与背景基因集,结果显示其在目标基因集中显著更高(如 ESR1 在 ATAC 和 H3K27ac 模块下的 p 值分别为 3.3e-38 和 8.6e-34)。随着预测排名下降,TR 的重要性逐渐减弱,而排名最末的 HDGF 表现出明显较低的调控潜能,说明 D-RP 能有效区分关键 TR 与非关键 TR。
此外,研究人员构建了高低 I-RP 分数的 TR 活性曲线,发现高分组的 TRs 对基因集的信号更强,进一步支持 TRAPT 的预测准确性。
考虑到 ESR1 可结合远端增强子(如 ERSE),TRAPT 利用其专属的调控潜能模型对远端调控进行建模。研究人员将增强子分为近端与远端两类,发现预测出的 TR 更倾向于在目标基因附近的增强子区域中结合,而非背景区域。GATA3 和 FOXA1 明显偏好近端区域,ESR1 与 EP300 则未表现出显著偏好。
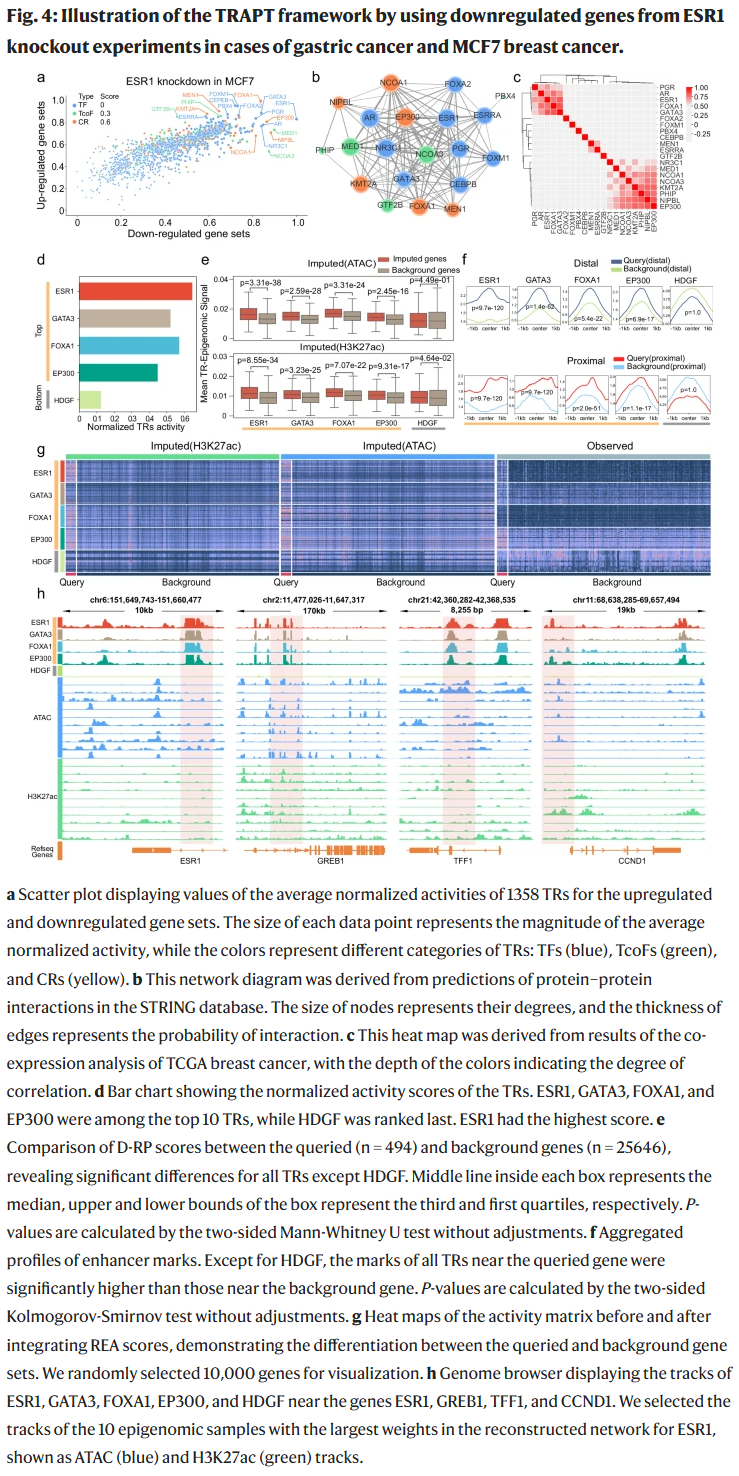
最后,研究人员可视化了 ESR1、GATA3、FOXA1、EP300 和 HDGF 附近的差异表达基因轨迹,发现前四者在目标区域均表现出明显的结合峰,而 HDGF 则缺乏这种特征。这一系列结果进一步验证了 TRAPT 在预测 TF、协同因子及染色质调控因子方面的可靠性和解释力。
预测阿尔茨海默病相关的关键调控因子
转录因子(TF)结合位点上的遗传变异可能改变其结合能力,进而影响基因表达和细胞功能。为验证 TRAPT 在疾病研究中的适用性,研究人员将其应用于阿尔茨海默病(AD)GWAS 后分析。研究人员利用 MAGMA 工具从 GWAS 汇总数据中获得 AD 相关基因集作为 TRAPT 的输入,并结合细致的精细定位与结合位点共定位分析,识别出一系列被致病变异影响的关键 TRs。
在欧洲人群 GWAS 数据的基础上,TRAPT 所预测的前 25 个 TRs 中,有 68.2% 的结合 SNP 来自 AD 因果变异(p = 2.5e-12),而排名后 25 个 TR 的比例显著更低,表明排名越高的 TR 越可能与 AD 密切相关。进一步分析发现,TRAPT 排名前列的 TRs(如 SPI1、RELA、REST)在致病变异上的结合强度高于背景变异。例如,SPI1 与 71 个致病变异发生交集,远高于背景(p = 3.6e-4)。GSEA 统计检验也证实前列 TRs 与因果位点显著富集。
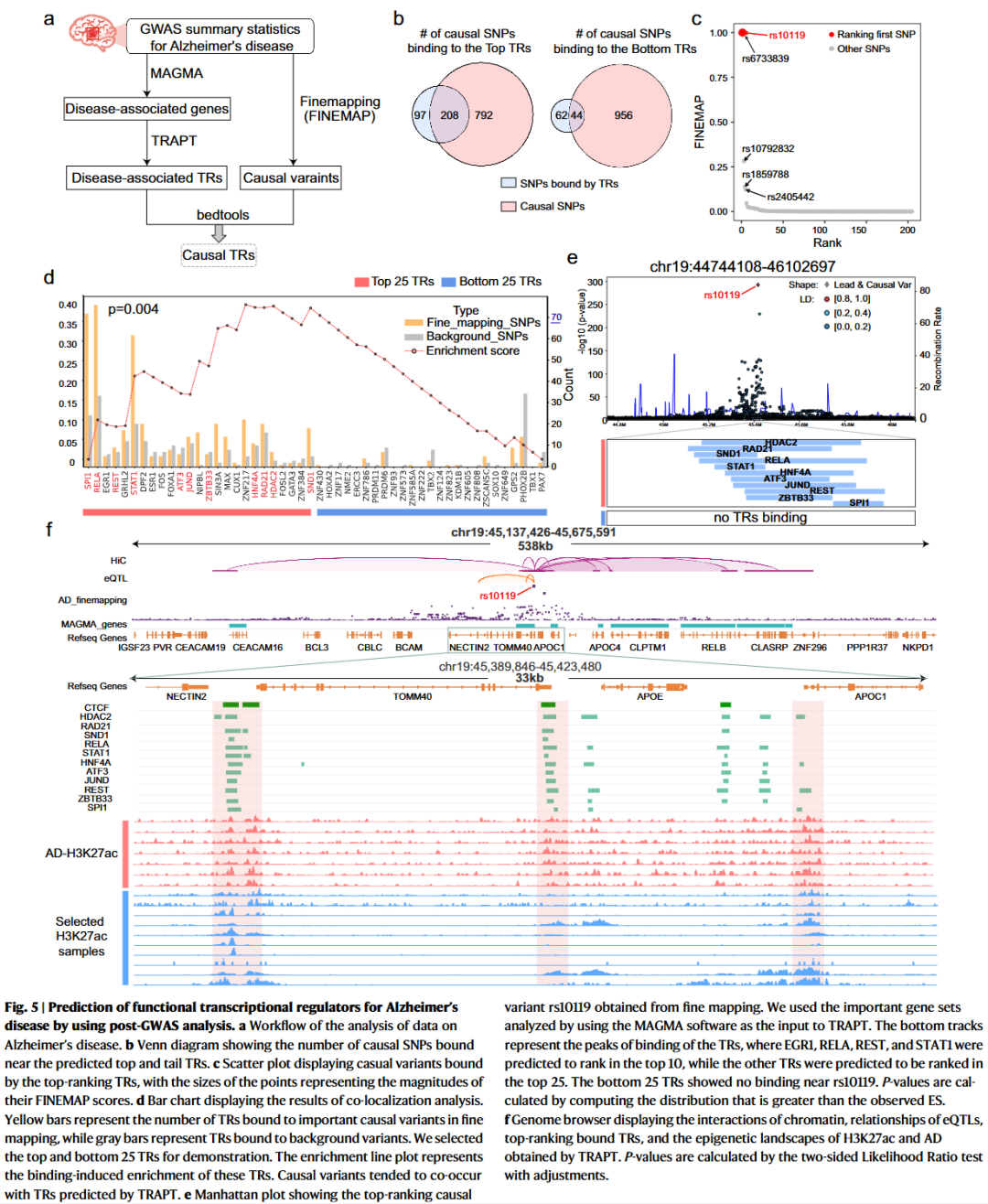
通过 FINEMAP 打分和功能注释,研究人员发现 rs10119 为评分最高的 AD 相关变异,与 APOE、TOMM40 和 APOC1 等关键基因相邻,且被多个预测的 TR 所结合(如 REST)。rs10119 位于染色质环结构的关键节点,多个已知 AD 相关的 TR(SPI1、STAT1、HDAC2 等)都在其上下游区域结合,进一步强化了其功能重要性。此外,在 AD 相关的 H3K27ac ChIP-seq 数据中,TRAPT 所选样本的调控区域与 rs10119 高度重叠,说明模型即便不依赖 AD 特异的表观数据,也能选择出具有相似信号的替代样本。
识别驱动细胞命运和组织特异性的关键调控因子
转录调控因子在细胞命运决定、分化过程和组织特性维持中起关键作用。研究人员进一步将 TRAPT 应用于单细胞转录组数据,以识别血液细胞分化路径中的驱动因子。在人类造血干细胞 scRNA-seq 数据中,TRAPT 成功识别了 42 个潜在关键 TR,其中 29 个已知与造血发育密切相关,10 个在不同分化亚群中呈现显著差异表达。
此外,部分 TRs 出现在多个谱系中(如 EP300、SMAD1、SPI1、TAL1),另一些如 STAT4 和 TCF4 则特异性分布于 NK 和 pDC 谱系,具有已知的调控功能。在另一个人类胚胎干细胞数据集中,TRAPT 同样成功识别出 GATA3、TFAP2A、GATA2 等在滋养层细胞中活性增强的 TRs,以及 GATA6、SMAD2 和 EOMES 等在内胚层细胞中活跃的 TRs,这些结果均与既往研究一致。
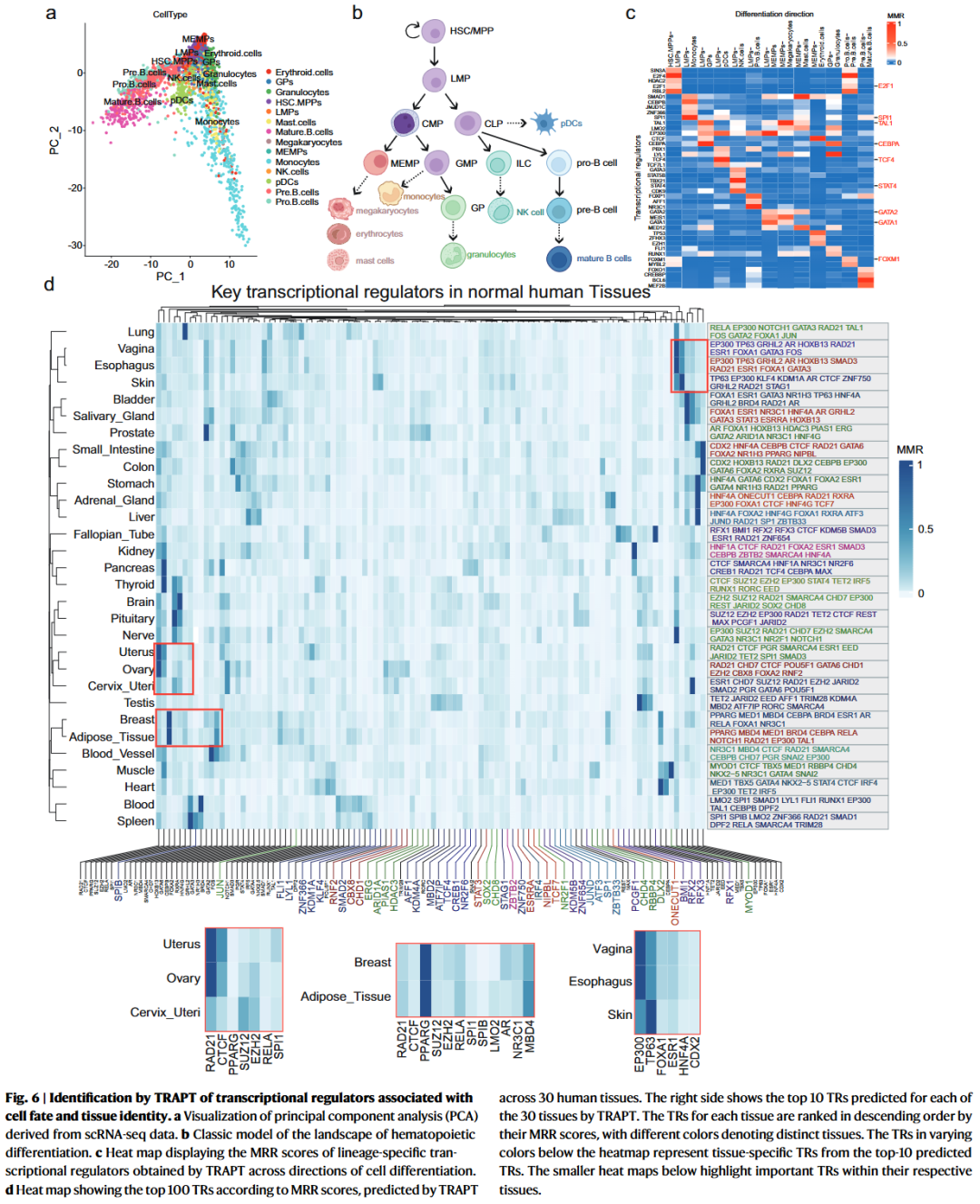
对 GTEx 项目中30种正常人组织的 RNA-seq 数据分析中,TRAPT 利用每个组织的差异表达基因成功预测出特异性调控因子。例如,心脏组织中识别出 MED1、TBX5、GATA4;前列腺中识别出 AR、FOXA1、HOXB13;而乳腺与脂肪组织共享了 PPARG 和 CEBPA。在对预测结果进行层次聚类时,乳腺与脂肪组织、女性生殖系统相关组织(如子宫、卵巢、宫颈)因细胞组成相似而聚类在一起,进一步验证了模型的生物学合理性。
讨论
转录调控因子(TRs)在调控基因表达、维持细胞稳态和引导发育过程中起着核心作用。TR 介导的转录程序通常具有独特的表观基因组图谱,并在细胞状态变化与疾病发生中充当“开关”。然而,当前针对特定基因集(如差异表达基因、单细胞标记基因)准确预测其上游TR仍面临挑战,主要受限于缺乏覆盖多样细胞类型的TR表观数据。
为解决这一问题,研究人员提出了深度学习框架 TRAPT,通过双阶段知识蒸馏提取调控元件的活性表示,整合超过 20,000 个大规模表观基因组样本与全面的TR背景知识库,精准预测上下文依赖的关键TR。在570个TR敲除/敲低数据集上,TRAPT 相较 Lisa、BART、i-cisTarget、ChEA3 等主流方法表现出显著性能提升,成功识别出与疾病、遗传变异、细胞命运与组织特异性密切相关的核心TRs。
当前TR预测方法主要分为两类:一类是基于基因集富集分析的方法,如 Enrichr、TFEA.ChIP、ChEA3 和 MAGIC,依赖统计检验评估TR的重要性,但无法模拟TR与顺式元件的真实结合;另一类如 i-cisTarget、BART 和 Lisa,通过模拟TR结合预测活性,但未能充分考虑TR的结合偏好。而 TRAPT 则提出第三种路径,融合基因集的顺式调控特征与 TR 的全基因组结合潜能,通过多阶段融合策略显著提升预测性能。
TRAPT 的优势包括:
融合式策略解决信息覆盖不全与结合偏好问题:多阶段建模同时考虑上下游调控潜能,填补以往方法中顺式图谱覆盖不全与TR结合偏好未建模的空白;
双阶段知识蒸馏增强模型鲁棒性:在 D-RP 阶段引入图变分自编码器(VGAE)与条件变分自编码器(CVAE)作为教师网络,缓解分布不一致问题;在 U-RP 阶段,蒸馏引导学生网络选取最优表观样本,提高数据整合能力;
图模型优化全基因组调控潜能预测:尤其适用于样本量有限的数据集,补全表观调控网络中的缺失连接,提高泛化能力。
消融实验表明,去除 D-RP、U-RP 或蒸馏机制均显著降低模型性能,验证这些模块的关键作用。同时,在连接预测任务中,D-RP 模型在多种掩码比例下表现稳定,展示出对缺失扰动的鲁棒性。
应用层面,TRAPT 成功识别了 ESR1 敲除实验中的 ESR1 及其相关的协同因子 EP300、FOXA1;在阿尔茨海默病研究中,TRAPT 能捕捉到如 SPI1、RELA、REST 等高可信TR与GWAS致病位点(如 rs10119)的共定位;在人类造血干细胞、胚胎干细胞及 GTEx 组织样本中,准确预测出与细胞命运(如 GATA3、STAT4、TCF4)和组织特异性(如 MED1、GATA4、AR)相关的关键TRs。所有结果均从多个角度验证了 TRAPT 在多类任务中的稳定性与通用性。
值得注意的是,尽管 TRAPT 整合了 17,227 个TR、1,329 份 ATAC-seq 与 1,465 份 H3K27ac 数据,但部分 TR 仍缺乏对应的表观样本,可能影响模型精度。同时,不同 TR 之间的协同或拮抗机制(如转录因子与协同因子之间的相互作用、染色质调控因子的辅助调控)在目前版本中尚未完全建模。未来可引入基因调控网络与因果推理策略,更系统地模拟调控层级间的复杂交互。
整理 | WJM
参考资料
Zhang, G., Song, C., Yin, M. et al. TRAPT: a multi-stage fused deep learning framework for predicting transcriptional regulators based on large-scale epigenomic data. Nat Commun 16, 3611 (2025).
https://doi.org/10.1038/s41467-025-58921-0
内容中包含的图片若涉及版权问题,请及时与我们联系删除


评论
沙发等你来抢