
DRUGAI
今天为大家介绍的是来自美国加利福尼亚大学,美国波士顿东北大学团队合作的一篇论文。虚拟库对接可以揭示与生物靶标结构互补的意外化学类型。在寻找大麻素-1受体(cannabinoid-1 receptor,CB1R)激动剂(agonists)的过程中,作者对7400万个实体分子进行对接,并优先选择46个高排名分子进行从头合成和测试。通过放射性配体竞争实验,其中9个显示活性,命中率为20%。基于结构的优化,对其中一个最有效的化合物(Ki = 0.7 μM)进行改进,得到了'1350,一个0.95 nM的配体,是Gi/o信号通路的完全CB1R激动剂。冷冻电镜结构显示'1350与CB1R-Gi1复合物的构象确认了预测的对接位置。这种先导激动剂在雄性小鼠中表现出强烈的镇痛作用,与运动减少、镇静和强直症状相比具有2-20倍的治疗窗口,且没有观察到条件性位置偏好。这些发现表明,独特的大麻素化学类型可能将特征性大麻素副作用与镇痛作用分离开来,支持进一步开发大麻素作为疼痛治疗药物。

尽管大麻素的治疗用途可追溯至至少15世纪,但其在现代治疗中的应用,例如作为镇痛剂,因其镇静和改变情绪的作用以及对其强化和成瘾潜力的担忧而发展缓慢。随着大麻法律地位的变化和减少对阿片类药物依赖于疼痛管理的努力,人们对理解内源性大麻素系统及如何利用它进行治疗开发的兴趣重新增加。此类治疗的潜在应用领域包括焦虑、恶心、肥胖、癫痫和疼痛,后者是本研究的重点。进展缓慢的原因包括大麻素本身的物理特性(通常高度疏水性)、不确定法律环境的挑战,以及药物常伴随的实质性不良副作用,包括镇静、精神作用,以及对强化和成瘾的担忧。事实上,大麻素的一个特征性定义特点是它们的"四联征":镇痛、体温降低、强直和运动减少,后三者可能被视为不良药物反应。同时,人体临床试验中的不确定结果导致该领域对大麻素作为治疗药物的有效性存在不确定性。尽管如此,对非阿片类镇痛剂的强烈兴趣,以及大麻素在动物疼痛模型中的明显功效仍然维持了对这些靶点的治疗兴趣。
大麻素-1和-2受体(CB1R和CB2R),均为G蛋白偶联受体(G protein coupled receptors,GPCRs)脂质家族成员,是大麻素活性的主要介导者。这两种相关受体主要通过其表达谱区分,CB1R在整个神经系统和身体中表达,而CB2R主要在外周免疫细胞中表达,尽管后者的确切分布仍是争论的主题。基于这些表达谱,并由动物研究支持,CB1R被认为是涉及大麻素精神活性和四联征效应的主要靶点,以及它们在疼痛测试中的镇痛作用,尽管由于两个受体的高度相似性和CB2R的外周分布,若没有直接测试,后者的作用常常不能被排除。
这些受体结构的确定提供了使用基于结构的方法寻找具有独特化学类型的配体的机会。最近基于结构对按需制造的虚拟库进行对接,已经为一系列靶点发现了这类分子,这些分子通常具有差异性药理学和减少的副作用。通过推广,从基于结构的方法中产生的额外CB1R化学类型可能解决当前大麻素的一些不足。
在这项工作中,作者对人类CB1R计算机对接了7400万个虚拟但易于获取的分子库,揭示了一系列具有相对有利物理特性的不同支架。基于结构的优化产生了具有低纳摩尔结合亲和力的激动剂。主要激动剂是一种强效镇痛剂,在低至0.05 mg/kg的剂量下具有镇痛活性。它在镇痛与运动减少、镇静和强直之间有2至20倍的分离,解决了四联征的多个负面方面,并突显了基于结构筛选在识别具有差异性药理学的化学支架方面的实用性。
针对CB1R的虚拟库对接
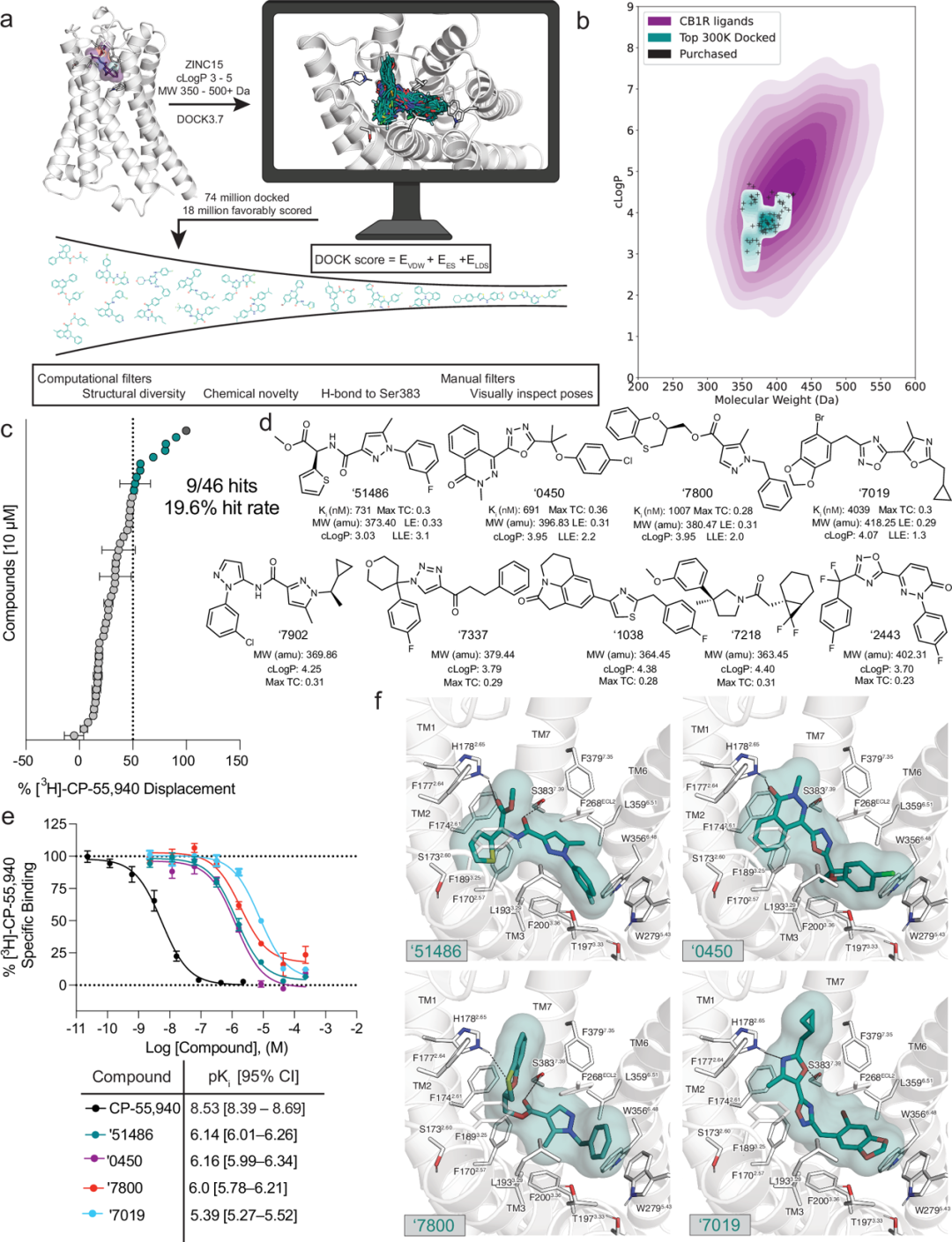
图 1
CB1R的正构位点既大又亲脂,这解释了其许多配体的高分子量和疏水性(图1)。然而,这些物理特性通常会带来代谢和溶解性挑战。为了平衡药物特性与补充CB1R位点所需的特性,作者从ZINC15数据库中含有7400万分子的"类先导物"子集中寻找分子,这些分子介于350至500原子质量单位之间,计算的LogP(cLogP)在3至5之间。这个范围与已知的CB1R特性空间重叠,同时保持了相对于许多大麻素的极性和尺寸优势(图1b)。每个分子以平均304万种位姿(方向×构象)进行对接,总共抽样和评分了63万亿个复合物。为了寻求多样化的候选物,排名前30万的分子被聚类为60,420组,并对每组得分最高的成员使用拓扑不相似性与ChEMBL中已知CB1/CB2受体配体进行筛选,采用拓扑系数(Tc <0.38)比较延伸连通性指纹最多至四个键(ECFP4)。与已知配体不相似的高排名化合物被筛选为与S3837.39和H1782.65可能的极性相互作用(上标表示Ballesteros-Weinstein命名法;见方法,图1a,表1)。剩余排名前1万的分子在UCSF Chimera中进行视觉评估,评估那些在评分函数和化学信息学筛选中未包含或近似的特征,如氢键的角度和距离、二面角应变,以及不正确的质子化或互变异构。最终,60个被优先考虑进行从头合成。其中,46个成功合成并测试了CB1R活性。与库的设计一致,这些分子比大多数现有大麻素配体更小且更极性,沿着适合CB1R正构口袋大而疏水的特性空间边缘(图1b)。
在单点放射性配体置换实验中,46个优先分子中有9个置换了超过50%的放射性配体,命中率为20%(图1c、d)。其中排名前四的分子(ZINC537551486、ZINC1341460450、ZINC749087800和ZINC518437019,以下分别简称为'51486、'0450、'7800和'7019)随后进行了完整的浓度-反应测试。这四个分子都能置换放射性配体3H-CP-55,940,Ki值范围从0.7到4 μM(图1e)。由于与抑制性Gαi G蛋白偶联,使用表达hCB1的细胞监测福斯可林(FSK)刺激的cAMP下降的功能有效性实验显示,'51486和'0450表现出适度的激动活性(图2a)。有限的溶解性阻碍了在足够高浓度下进行测试以获得准确的EC50值;幸运的是,胶体聚集物反筛选显示在10 μM以下没有这种活性(图2b-e),表明在结合和功能测定中看到的活性不是由于这种混杂因素所致。总的来说,这九个活性分子探索了一系列在拓扑上与已知CB1配体不同的化学类型,具有相对有利的物理特性(图1b、d)。
虽然识别出的配体在化学和物理方面与已建立的大麻素不同,但它们的对接姿态重现了已知配体的相互作用,只是使用了不同的支架和识别元素。四种最有效的配体对接时都采取了"C"形构象,这是MDMB-Fubinaca、AM11542和AM841与CB1R结合时实验观察到的特征几何形状。同样,所有四种配体预计都会与S3837.39形成氢键,这是几乎所有激动剂结合的配体-受体复合物中观察到的CB1受体决定效力的相互作用。此外,所有四种配体预计会与H1782.65形成次级氢键,这一特征被认为对CB1R的效力和激动作用也很重要。这些极性相互作用主要是通过独特的氢键受体基团形成的,如噁唑、噁噻因或吡啶酮。其他特征性疏水和芳香堆积相互作用遍布配体中,包括与F268ECL2、W2795.43和F1742.61的相互作用,尽管与已知配体相比,这些相互作用通常使用不同的芳香基团(图1f)。两个最有效的命中物('51486和'0450)进一步展示了与双切换残基W3566.48和F2003.36的芳香堆积和疏水包装,这些残基对受体激活很重要,可能解释了它们相比'7019和'7800更强的激动剂特性,虽然靶向效力也可能起作用。
为了优化这些初始配体,作者在包含120亿个可实现分子的库中,使用SmallWorld(NextMove Software)寻找与四个活性分子ECFP4 Tcs ≥ 0.5的分子。这些类似物按照与原始对接活动相同的标准进行构建、对接、筛选和选择。对每个支架合成了11至30个类似物。在四个初始命中物中,三个找到了优化的类似物,亲和力提高了5至24倍,其中'7019提高了5倍至Ki值87 nM,'0450提高了24倍至163 nM,'51486提高了16倍至44 nM(表2)。基于第一轮亲和力的改进,只有'51486系列进入了第二轮类似物开发。
在随后的专门合成(即非常规库合成)中,'51486的第一轮44 nM类似物'60154,在手性中心添加甲基后进一步优化为Z8504214042('4042),Ki值为1.9 nM。由于'4042是消旋体,作者使用手性色谱法将其纯化为其组分异构体Z8526711350('1350)和Z8526708690('8690),并测量CB1R结合以确定活性对映异构体。Ki值分别为0.95 nM和90 nM,'1350明显比其对映异构体更有效,随后的功能研究表明它是更强的激动剂。在努力去除'1350中的手性中心和潜在代谢不稳定性过程中,作者用二呋喃基取代物替换了'1350的甲基酯和噻吩,从而发现Z8703004936('4936),Ki值为7.5 nM。图2总结了整个'51486系列的结构-活性关系(SAR)和对接模型。
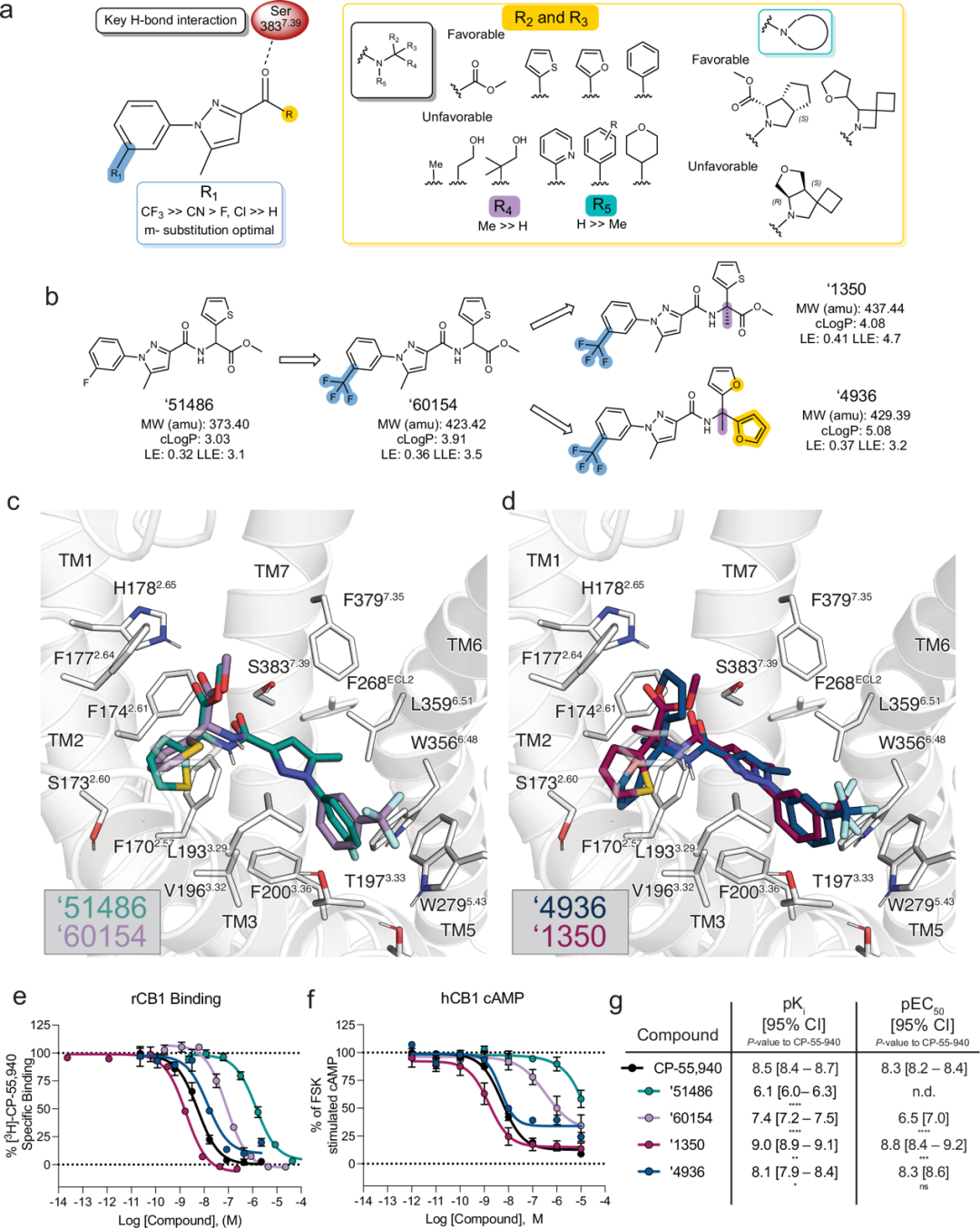
图 2
从结构-活性关系(SAR)中得到的关键经验包括R1位置体积大且疏水性基团的重要性,该位置被模拟为与W2795.43和T1973.33堆积(图2c),其中间位取代的CF3最为有利。R2和R3的取代对配体效力有不同影响,最有利的功能基团包括酯/噻吩('51486、'60154、'1350)、双呋喃('4936、'1090)或呋喃/苯基('5806、'1081)对。'1350的酯羰基和'4936的呋喃基氧被模拟为与受体的H1782.65形成氢键,尽管距离表明可能是水介导的相互作用或弱氢键(图2d)。与对接模型一致,酯的羧酸盐类似物'4051仅弱结合(Ki = 5 μM,效力低5,000倍)。此外,手性中心的甲基化(R4位置,'1350、'4936、'5806)显著改善了亲和力('4936约6倍,'5806约15倍,'1350约50倍;图2e)。这一添加预计会增加配体与跨膜螺旋2之间的范德华相互作用。相比之下,R5位置酰胺氮的甲基化('1066、'4388和'1082)使支架亲和力至少降低100倍,最多降低2000倍,尽管对接模型预测这是一个未满足的氢键供体。虽然作者没有尝试同时甲基化酰胺氮和手性中心(向'1350、'4936或'5806添加N-Me),但预计这也会对结合产生负面影响。鉴于结构相似性和效力差异,'4051、'1066和'4388可用作未来研究中的无活性探针对。
产生的先导化合物'1350和'4936都是CB1R的强效结合物,其中'1350为0.95 nM,比广泛使用的CB1R探针CP-55,940强3倍(P = 0.007),而'4936为7 nM,比CP-55,940弱2.5倍(P = 0.04)(图2e、g)。尽管'1350和'4936都比初始对接命中物'51486更疏水,但'1350的亲脂配体效率(LLE; LLE = pIC50 − clogP)从3.1提高到4.7(图2b),而'4936的LLE基本保持不变(3.2);这两者与阳性对照CP-55,940的LLE值2.6相比都更为有利(cLogP = 5.66),后者比这两种对接衍生的激动剂更疏水。
激动活性与亚型选择性
鉴于'1350和'4936(图2e)及其多个类似物具有较强的亲和力,作者接下来研究了它们与广泛研究的大麻素CP-55,940相比的功能活性。作者首先通过Lance Ultra cAMP测定法测量了Gi/o介导的激动活性,该方法通过抑制forskolin刺激的cAMP实现(方法部分)。在表达人CB1受体(hCB1R)的细胞中,'1350、'4936及其类似物都表现为激动剂,其EC50值与它们的亲和力相当(图2f、g)。这一系列中的大多数分子接近于完全激动剂,其最大效应(Emax)通常大于75%。唯一的例外是'4936,其Emax为65%,更符合强效部分激动剂的特征(图2f、g)。为了验证活性的可重复性,作者在正交G蛋白、β-arrestin-2以及脱靶测定中进行了进一步研究。
基于这种强效活性,并为了控制系统偏差,作者利用ebBRET bioSens-All®平台研究了'1350对CB1R和CB2R的多种G蛋白和β-arrestin-2("信号偏向性")的差异性募集。作者通过描述'1350对各信号通路的相对有效性(相对功效=10^{Δlog(Emax/EC50)})将其活性与CP55,940进行了比较。在CB1R中,'1350在募集Gi/o和G13亚型方面的相对功效约为CP-55,940的2倍,尽管募集的效应物模式相似。值得注意的是,'1350对G13和β-arrestin-2的最大效应(Emax)有所降低,这表明它对这些通路可能是部分激动而非完全激动,这可能具有一定的生理相关性。对于高度相关的CB2R,差异性活性在质上有所不同,与CP-55,940相比,'1350在所有募集的效应物中对CB2R一致表现为相对功效较低的部分激动剂。总之,'1350没有表现出强烈的功能选择性或偏向性,但在CB1R(而非CB2R)上比CP-55,940更有效且相对功效更高。
'1350-CB1R-Gi1复合物的冷冻电镜结构
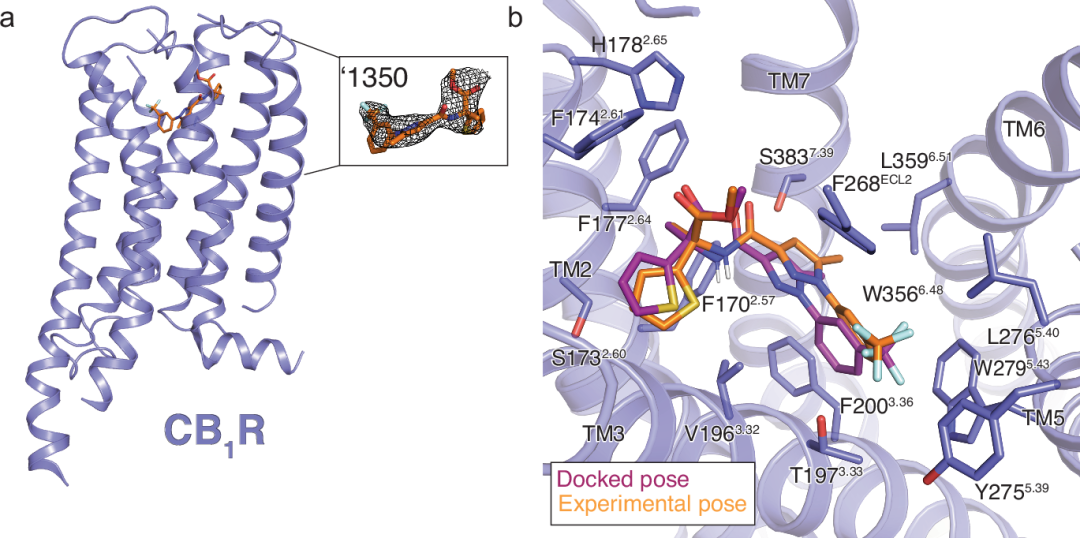
图 3
为了在原子分辨率水平上理解'1350的构效关系,并为未来的优化提供模板,作者测定了'1350-CB1R-Gi1复合物的冷冻电镜结构(图3),标称分辨率为3.3 Å。与先前人类CB1R激活状态的结构一致,配体占据了由跨膜螺旋(TMs)2-3和5-7形成的正交口袋,并由ECL 2覆盖。
'1350的实验结构与对接预测的R-对映体构象吻合良好,这一对映体对受体的对接得分更佳(R-对映体的DOCK3.7得分为-43,而S-对映体为-38)。在光旋转研究中,'1350被证明是(-)对映体(见补充信息文件),综合表明'1350是R/(-)对映体。预测和实验结构的全原子RMSD叠合度为0.78 Å(图3b)。尽管局部分辨率限制使得无法明确建模所有旋转异构体,但电子密度表明对接预测的与CB1R的主要相互作用可能在实验结构中得到保留,包括配体的酰胺羰基与S3837.39之间的关键氢键。三氟甲基与W2795.43和T1973.33残基形成范德华力和四极相互作用,这与对接结构预测一致,并且与亲和力提高1.7 kcal/mol('51486 Ki = 731 nM vs. '60154 Ki = 44 nM,仅添加CF3就使Ki提高了17倍)相符,这是由于用三氟甲基替代了原始的氟原子。
编译|黄海涛
审稿|王梓旭
参考资料
Tummino, T. A., Iliopoulos-Tsoutsouvas, C., Braz, J. M., O’Brien, E. S., Stein, R. M., Craik, V., ... & Shoichet, B. K. (2025). Virtual library docking for cannabinoid-1 receptor agonists with reduced side effects. Nature Communications, 16(1), 2237.
内容中包含的图片若涉及版权问题,请及时与我们联系删除

评论
沙发等你来抢