
DRUGAI
今天为大家介绍的是来自美国杜兰大学医学院的Tony Ye Hu与Wenshu Zheng团队发表的一篇论文。科研人员开发出一种新的群体关联模型(Group Association Model, GAM),用于发现细菌对抗生素产生抗药性的遗传变异特征。这个模型不需要依赖已有的专业知识库,就能准确找出与耐药性相关的基因变异,同时还能减少错误判断细菌对多种药物同时产生抗性的情况。研究团队用这个模型分析了超过7000株结核分枝杆菌(Mycobacterium tuberculosis, Mtb)样本。结果显示,与世界卫生组织(WHO)现有的基于专家经验的突变检测方法相比,GAM在识别各类药物的目标基因时表现相当,但更少出现误判。为了证明模型的通用性,研究人员还分析了近4000株金黄色葡萄球菌(S. aureus)样本,同样取得了很高的预测准确率。通过引入机器学习(ML)技术对GAM进行优化,模型在处理小规模或不完整数据时的预测能力得到进一步提升。研究团队在三个不同地点收集的427个结核分枝杆菌样本中验证了这一发现,结果表明GAM提供的数据比WHO的方法更适合用于机器学习预测。这种结合了GAM和机器学习的新方法有望突破现有耐药性预测技术的限制,为耐药性细菌感染的治疗方案选择提供更好的指导。

微生物耐药性已成为一个日益严重的全球健康问题。这种耐药性主要源于基因突变、基因横向转移以及抗生素的过度使用。最新研究显示,2021年全球新增了超过1000万例结核病病例,其中约160万人不幸死亡。特别值得注意的是,有45万例患者对常用的抗结核药物利福平产生了耐药性。虽然结核病的整体治疗成功率达到86%,但对于出现耐药性的病例,治愈率仅为57%。为了应对这一挑战,研究人员开发出一种新的群体关联模型(GAM)。这个模型通过分析7179个结核分枝杆菌样本,成功识别出8种主要抗结核药物各自对应的特定耐药基因,避免了传统基因组关联研究(GWAS)方法容易出现的错误关联。研究团队还用近4000个金黄色葡萄球菌样本验证了该模型的普适性。
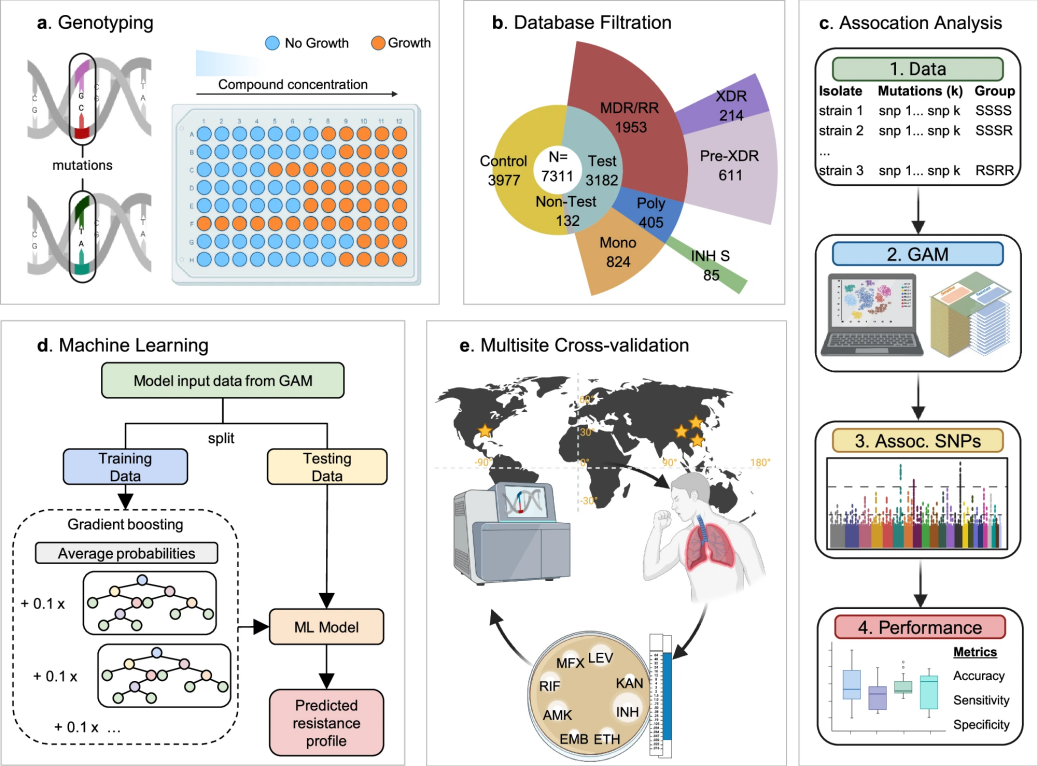
图 1
传统的耐药性检测方法存在明显缺陷:培养法不仅耗时费力,对于生长缓慢的细菌可能需要数周时间;分子检测方法虽然较快,但只能识别已知的常见突变;DNA测序虽然能发现新的突变,但准确判断这些突变是否导致耐药性仍然困难。研究发现,将GAM与机器学习技术相结合(如图1所示)能显著提升预测准确性,特别是在处理小规模或不完整数据时表现更为出色。这一发现为临床上快速识别耐药性细菌,制定更有效的治疗方案提供了新的可能。
耐药分离株的群体特征
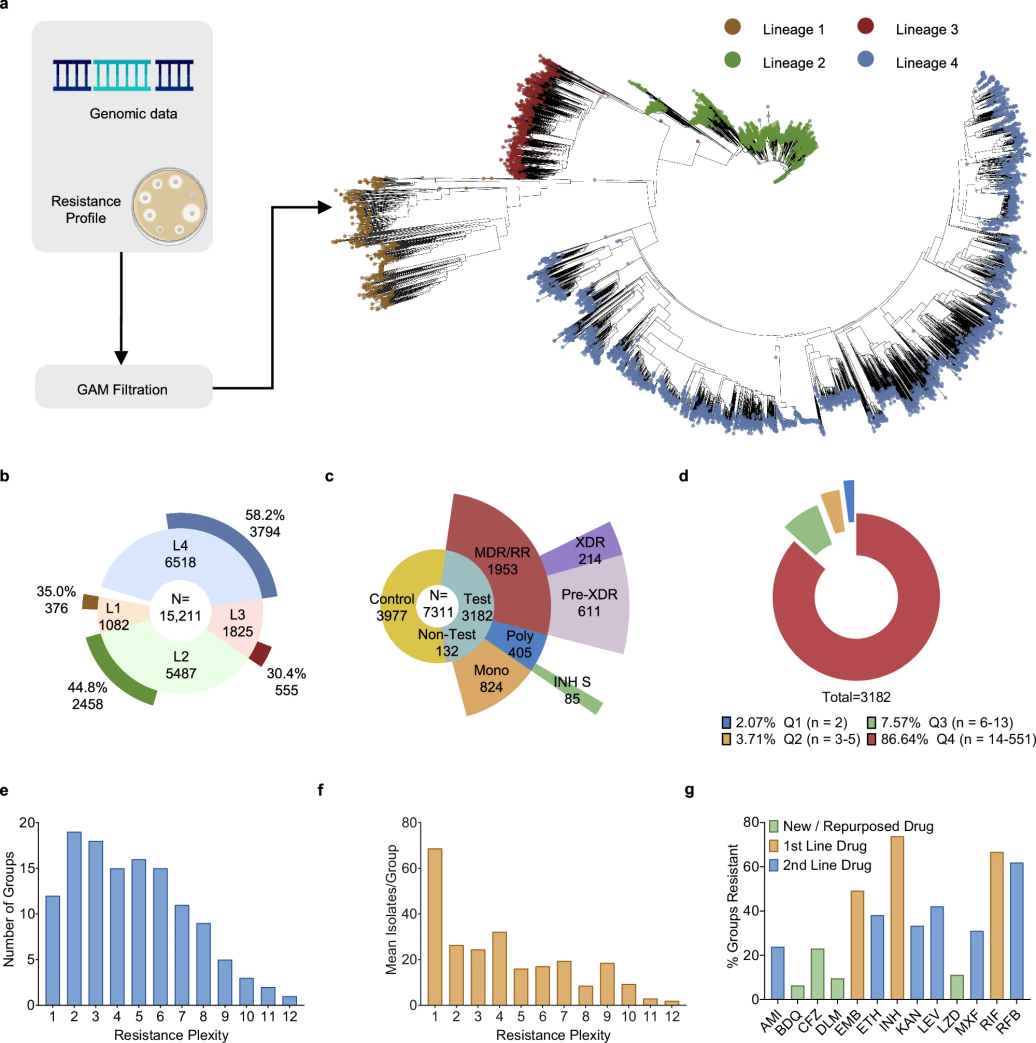
图 2
研究团队对超过1.2万个结核分枝杆菌基因组数据进行了全面分析。为确保研究质量,研究人员首先进行了严格的数据筛选,最终保留了约1万个高质量样本(DS1),其中包括7179个完整的耐药性数据样本(DS2)。通过系统发育分析(如图2a, b所示),研究人员发现这些样本在结核分枝杆菌的四个主要家族中分布均匀,这说明研究结果不会因为样本选择偏差而失真。为了更好地研究耐药性特征,研究团队创新性地将样本按照它们对不同药物的耐药模式进行分组,形成了126个研究组(如图2c所示)。
分析结果显示,在所有样本中,超过一半(54.7%)对13种受试药物都保持敏感。在耐药样本中,绝大多数(86.6%)属于较大规模的组别(每组至少14个样本)。研究还发现不同类型药物的耐药情况差异显著:一线用药中,异烟肼的耐药率最高,达到74%;其次是利福平,为67%;乙胺丁醇约为49%;二线替代药物利福布汀的耐药率也达到62%,其他二线药物的耐药率在24-42%之间;新型药物和重新启用的老药耐药率最低,仅为6-23%。这种分组分析方法不仅帮助研究人员更准确地识别药物与基因之间的关联,也为了解不同药物的耐药发展趋势提供了重要参考。
与耐药表型相关的基因和突变的鉴定
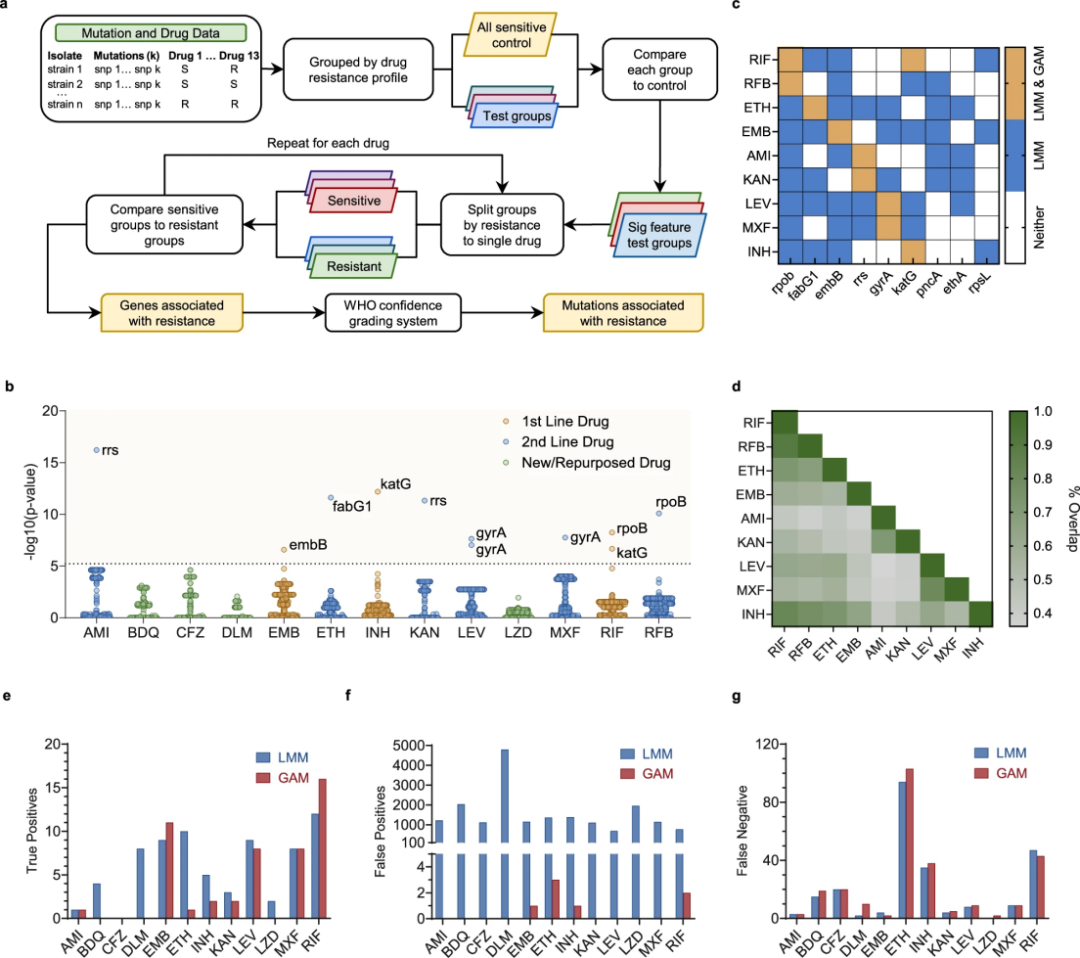
图 3
研究团队开发的GAM方法在识别耐药性相关基因变异方面取得了重要突破。如图3所示,这种新方法能够准确找出6个与主要抗结核药物相关的关键基因,且几乎不会出现误判,仅在异烟肼相关基因katG与利福平的关联中出现了一次误报。与传统的基因组关联分析方法(GWAS线性混合模型)相比,GAM的优势非常明显:
准确性大幅提升:传统方法在识别每种药物的目标基因时,往往会产生数千个错误关联,而GAM最多只有1-2个误判;
预测效能显著提高:GAM的阳性预测准确率是传统方法的200多倍;
抗干扰能力强:即使在不同耐药性之间存在重叠的情况下,GAM也能准确区分各种药物的特定耐药基因。
为了验证GAM的通用性,研究人员还分析了近4000个金黄色葡萄球菌样本。结果表明,GAM不仅能识别出由基因突变引起的耐药性(如环丙沙星等3种药物),还能发现通过基因在细菌之间传递而产生的耐药性(如庆大霉素等4种药物)。这些发现对临床治疗具有重要意义:GAM能够帮助医生更准确地预测病原体对不同抗生素的耐药性,从而制定更有效的治疗方案,减少抗生素的滥用,延缓耐药性的发展。
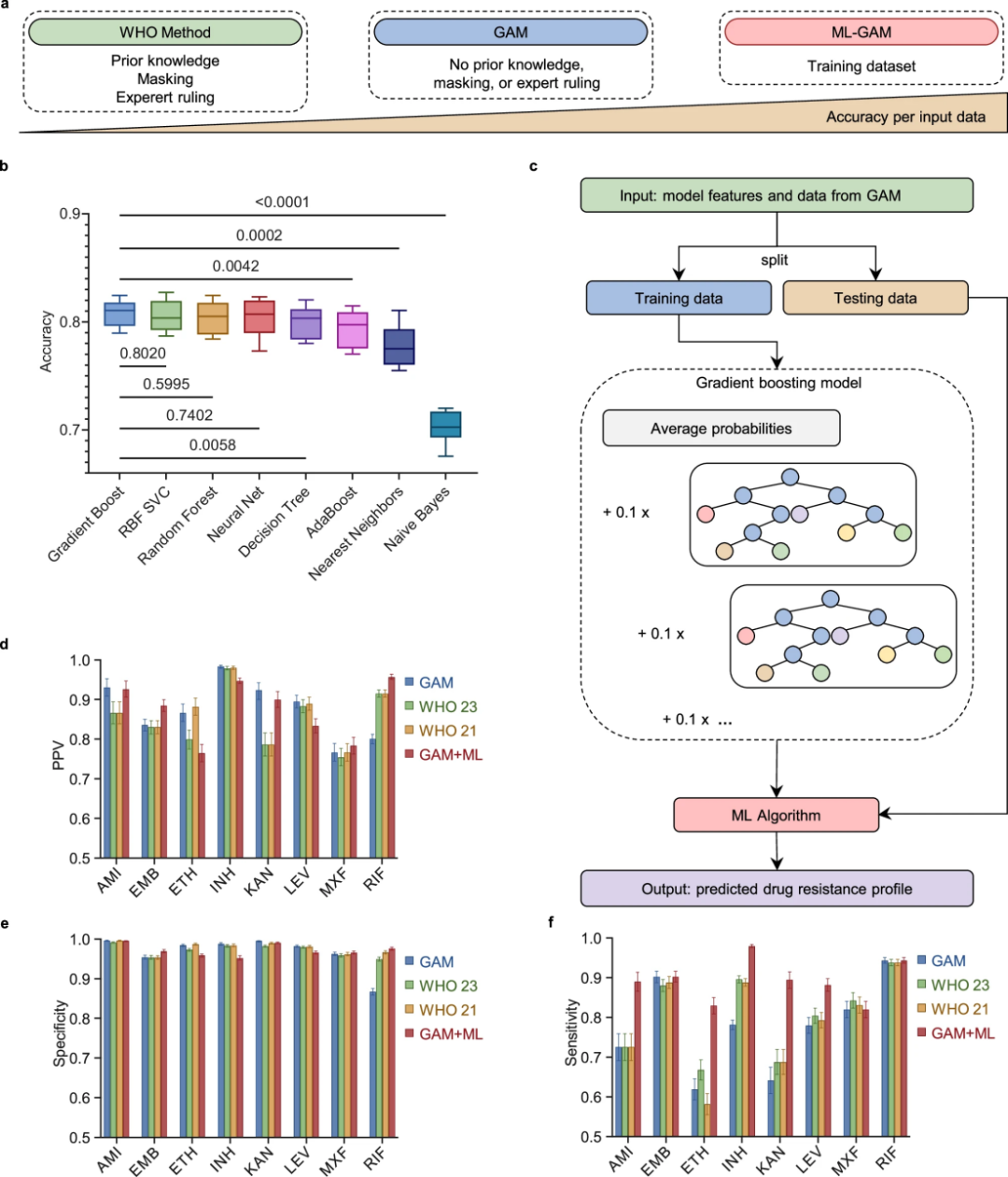
图 4
如图4所示,研究团队比较了GAM、WHO目录和GAM+机器学习三种方法的预测性能。GAM与WHO的2021和2023年结核分枝杆菌突变目录的重叠率为1.2-80.0%。在9种机器学习模型中,梯度提升法表现最佳,平均准确率达81.0%,方差最低(1.66%)。GAM+机器学习组合在预测利福平耐药性时,特异性提高了10.9%。对于阿米卡星、乙硫异烟胺、异烟肼、卡那霉素和左氧氟沙星等药物的敏感性也有所提升。接收者操作特征曲线(AUC)值达到85.0-97.0%,优化后的模型整体准确率提高了2.6%。
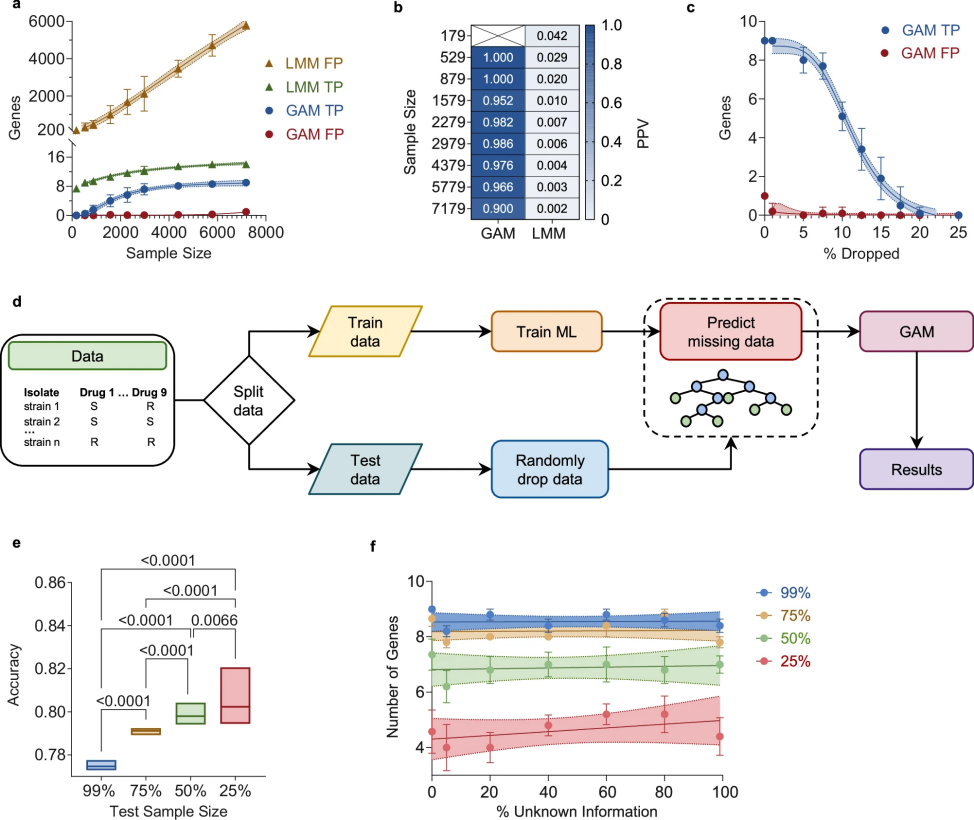
图 5
在图5中,研究团队评估了样本量和数据完整性对GAM性能的影响。在179-7179个样本的测试中,GAM与样本量呈渐近关系,在中点处出现拐点。虽然LMM识别出的真阳性略多,但其假阳性率是GAM的100-1000倍。数据缺失率与基因识别准确性呈反S形关系。通过机器学习优化后,即使只用1%的数据作为训练集,预测准确率也能达到0.767,接近使用75%数据的模型(0.801)。这说明机器学习能够有效缓解数据缺失对GAM分析的影响,但GAM的基因检测仍然依赖于样本量。
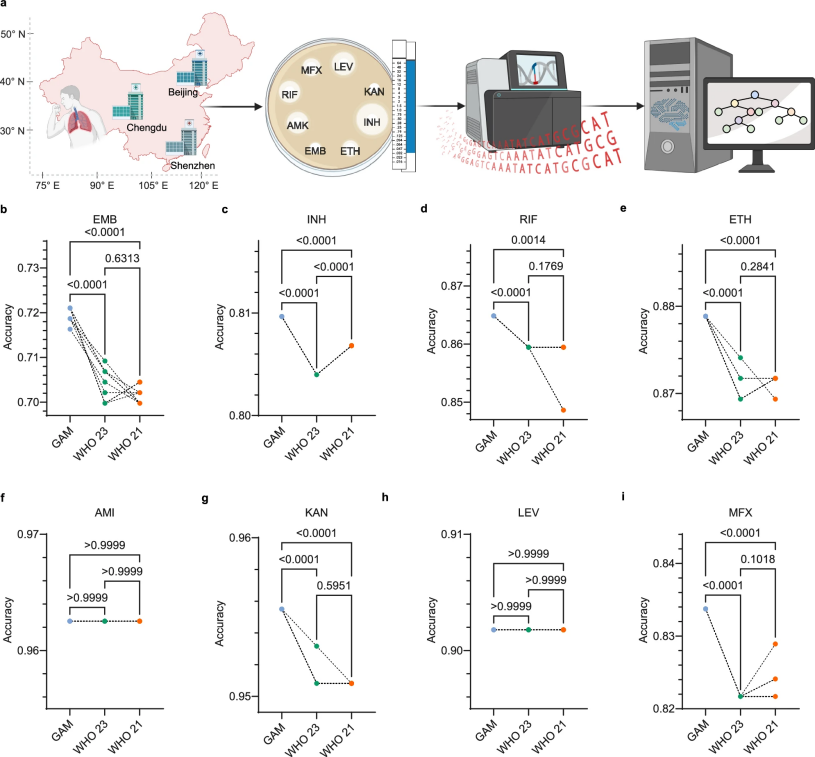
图 6
随后,研究团队比较了GAM和WHO目录在机器学习预测中的表现(图6)。使用梯度提升机器学习模型测试发现,GAM在预测阿米卡星、异烟肼、卡那霉素和利福平的耐药性时,准确率高于WHO模型。在427个来自中国三家医院的结核分枝杆菌样本验证中,GAM+机器学习模型在预测6种药物(乙胺丁醇、乙硫异烟胺、异烟肼、卡那霉素、莫西沙星和利福平)的耐药性时表现更优,在其他药物上的预测效果持平。
讨论
研究团队开发的GAM方法在分析复杂耐药谱的微生物群体时展现出独特优势。与传统方法相比,GAM不依赖已知的耐药机制知识,能自动过滤中性突变,减少假阳性和交叉耐药性的干扰。特别是当与机器学习技术结合后,GAM在处理完整数据和不完整数据时都表现出色,为预测药物-基因关联提供了更准确的工具。虽然GAM在识别水平基因转移等某些耐药机制方面还有局限,但其在多种病原体(如结核分枝杆菌、金黄色葡萄球菌等)的耐药性分析中都展现出良好应用前景。这一方法不仅可用于指导临床用药,还可能帮助作物育种等其他领域的抗性研究。
编译|于洲
审稿|王梓旭
参考资料
Saliba J G, Zheng W, Shu Q, et al. Enhanced diagnosis of multi-drug-resistant microbes using group association modeling and machine learning[J]. Nature Communications, 2025, 16(1): 2933.
内容中包含的图片若涉及版权问题,请及时与我们联系删除

评论
沙发等你来抢