
DRUGAI
今天为大家介绍的是来自美国约翰霍普金斯大学Jeffrey J. Gray团队的一篇论文。在蛋白质中,任何位置上的最佳残基是由其结构、进化和功能环境决定的——这很像语言中如何可以从上下文推断出一个词。作者训练了掩码标签预测模型,以学习不同环境中氨基酸残基的表示。作者关注进化和结构灵活性的问题,以及通过预训练和微调获得的上下文编码是否以及如何改进专门环境的表示。从作者学习的表示中采样的序列可以折叠成模板结构,并反映了相关蛋白质中观察到的序列变异。对于灵活的蛋白质,采样序列可以遍历天然序列的完整构象空间,这表明可塑性已经编码在模板结构中。对于蛋白质-蛋白质界面,生成的序列在体外模拟中复制了野生型结合能量,跨越了各种界面和结合强度。对于抗体-抗原界面,微调重现了保守的序列模式,而在一般环境下的预训练改善了超可变H3环的序列恢复。

近年来,已有多种深度学习模型被提出用于结构条件下的蛋白质序列设计,这些模型在各种设计任务中取得了成功。然而,目前尚不清楚基于结构条件生成的序列与来自进化家族的序列相比如何。对于具有多种构象的蛋白质,生成的序列范围是否也会表现出多种构象?此外,对于特定的进化环境,例如数据丰富的抗体或数据稀疏的T细胞受体,特定环境模型相比一般蛋白质模型表现如何?或者,何时训练一般蛋白质机器学习模型最佳,何时训练特定进化环境的模型最佳?为解答这些开放性问题,作者训练了掩码标签预测模型,以学习不同结构、功能和进化环境中氨基酸残基的表示。作者关注进化和结构灵活性的问题,以及通过预训练和微调获得的上下文编码是否以及如何改进专门环境(如抗体-抗原界面)的表示。由于篇幅限制,此处仅展示部分内容。
模型部分
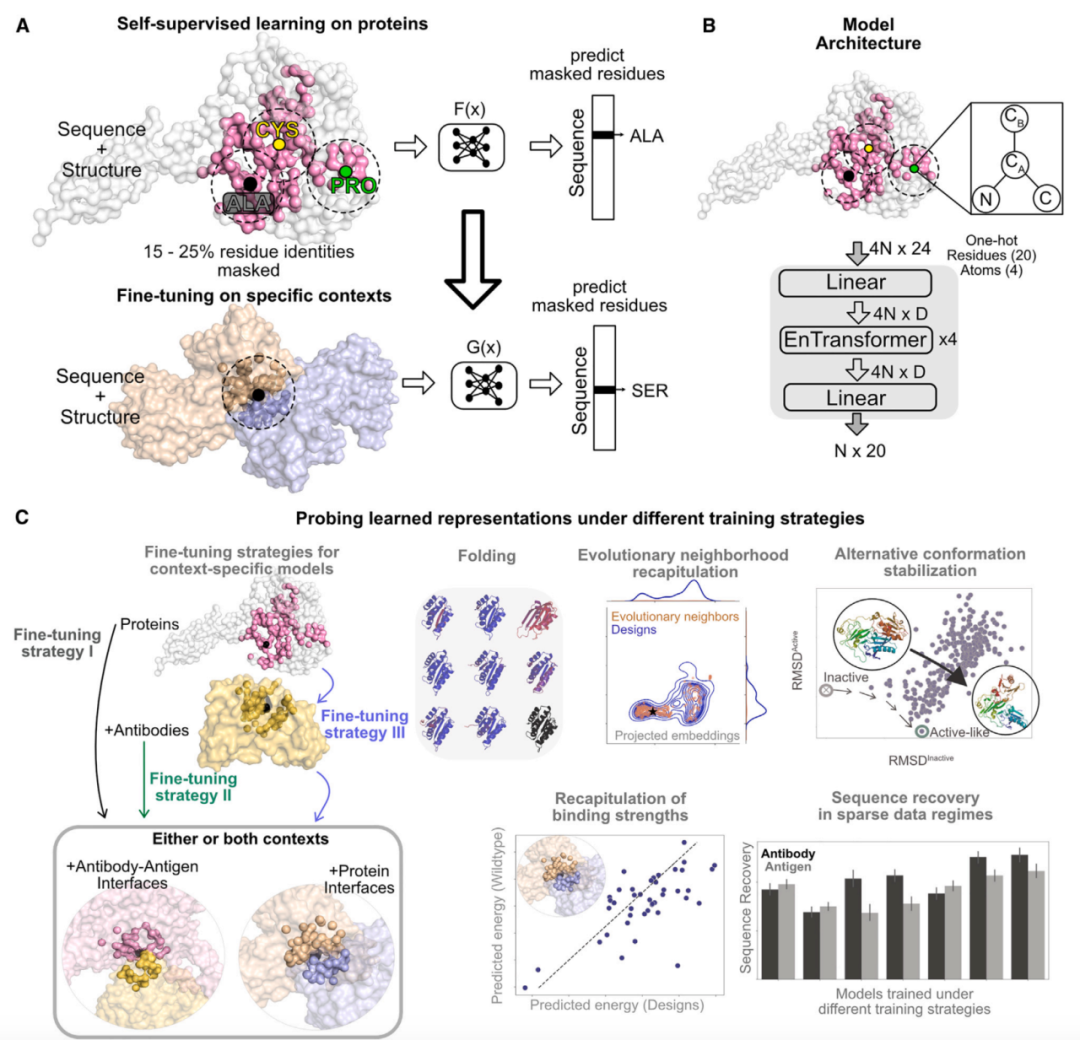
图 1
在自然语言处理中,词的语义可能来源于它出现的上下文。自监督技术如掩码语言建模(MLM)学习词在其上下文中的表示,这些表示可泛化到各种自然语言任务。在MLM中,基于注意力的transformer模型被训练来预测句子中缺失的词。模型通过随机掩盖一小部分词进行训练,并学习基于序列中非掩码词提供的上下文来预测被掩码的词。UniMP,一种用于图结构数据的掩码标签预测模型,模拟了在图中从已标记样本向未标记样本传递标签信息的过程。作者将MLM和UniMP的概念框架应用于从非掩码氨基酸残基在其结构和序列环境中传递氨基酸残基标签的学习,到在其结构和部分或完全掩码序列环境中的掩码氨基酸残基(图1A)。
作者的模型受到BERT多层双向transformer编码器的启发(图1B)。作者的编码器由四个基于等变图神经网络(EGNN)的E(n)transformer块组成,并修改为包含类transformer层,用于基于注意力的邻近节点聚合、前馈层和多头注意力。每个残基由三个主链原子(N、CA和C)和一个侧链原子(CB)表示。节点特征包括二十个独热编码序列标签(氨基酸残基)和四个独热编码原子标签(N、CA、C和CB)。所有氨基酸残基,包括甘氨酸,都通过这种表示得到适当表示(甘氨酸的CB编码为0)。对于掩码残基,CB被移除(独热原子编码设为零,与甘氨酸相同),其坐标被替换为零。空间中的相对位置和序列中的残基位置分别用Roformer和正弦嵌入编码。当存在多条链时,位置索引偏移100个残基。每个原子连接到其48个最近邻原子。作者使用256维的隐藏维度(D),分布在8个注意力头上。本工作中描述的所有模型有4.2百万个参数。
首先,作者在PDB数据集中的单链蛋白质上用MLM目标训练了一个通用蛋白质模型Prot_EnT,该数据集按50%聚类处理(有关训练集的详细信息,请参见STAR方法)。在最多15%部分掩码的训练后,该模型在低掩码率(15%)时可以恢复高达45%的序列一致性,并且在100%掩码时仍能恢复37.4%的序列一致性。作者还确认调整采样温度允许作者使生成的序列从低温下的最大似然序列变化到高温下更多样化的序列。作者探索了架构变化(图S2)并发现具有4个块的模型性能最佳。作者还确定在训练期间改变种子会导致序列恢复率的基线变化为4%,困惑度的基线变化为1.3比特。接下来,作者研究在100%掩码率下从Prot_EnT模型采样的蛋白质序列。
生成的蛋白质序列折叠成目标结构并采样附近的结构空间
作者首先检查了生成的序列在其预测结构中如何变化,以及这种变化如何在不同蛋白质间转变。使用TS50测试集,作者为50个蛋白质生成了最大似然设计。在这些蛋白质中,43个目标设计折叠成与天然结构的均方根偏差(RMSD)在2埃以内的结构。在最大似然设计未能折叠到天然结构的七个目标中,作者观察到AF2也未能将其中一个目标的野生型序列折叠到其天然结构。对于剩余的六个目标,作者采样了50个设计并计算了AF2折叠结构相对于天然结构的RMSD。采样序列在六个目标中只有三个目标能够得到与天然结构RMSD在2埃以内的折叠结构。因此,虽然Prot_EnT可以为49个目标中的46个(忽略AF2无法折叠野生型序列的一个目标)采样出能折叠成目标结构的序列,但仅编码器的Prot_EnT无法为TS50集合中至少3个目标采样出能折叠成目标结构的序列。这表明仅编码器模型可能不够,而迭代解码对于正确设计某些蛋白质结构可能至关重要。
在最大似然设计折叠成目标结构的43个目标中,作者选择了两个蛋白质进行进一步分析——一个具有高最大似然序列恢复率49%(甘露糖转运蛋白;PDB:1PDO)和另一个具有低最大似然序列恢复率27%(抗血小板水蛭蛋白;PDB:1I8N)。
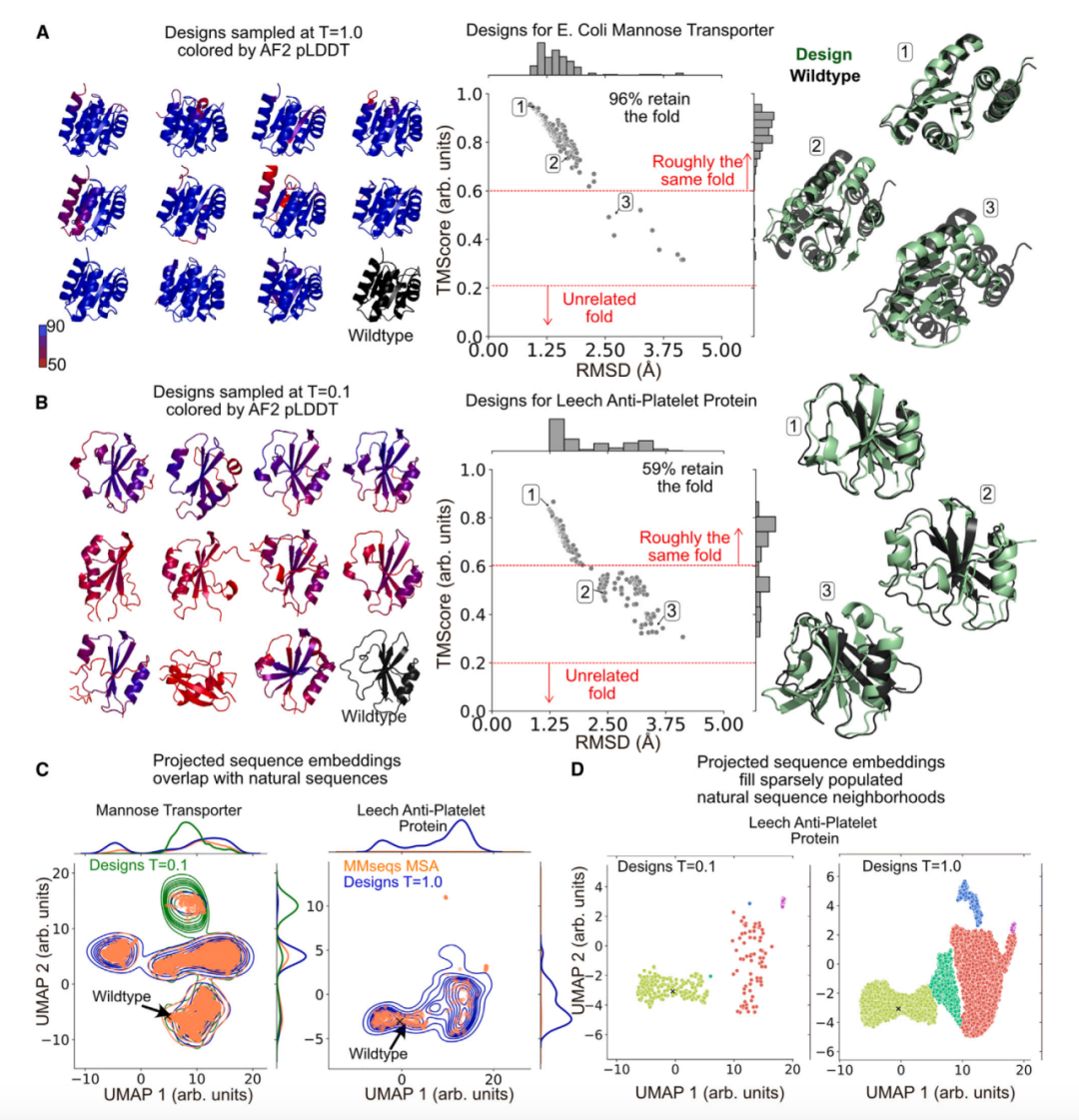
图 2
作者为每个蛋白质基于晶体结构采样了200个序列,并用AF2对它们进行折叠。对于甘露糖转运蛋白,96%的结构保持了相同的折叠(模板建模评分或TM评分≥0.5)并表现出总体良好的pLDDT(预测的局部距离差异测试)值(图2A)。对于抗血小板水蛭蛋白,只有59%保持相同的折叠,pLDDT评分较低(图2B)。作者检查了与原始结构具有低和高RMSD的代表性结构(图2A和2B中选定的结构1、2和3)。具有较高RMSD的结构("2"和"3")捕获了起始结构中的许多结构基序,但在α-螺旋排列或β链的数量上有所不同。这表明对给定结构采样的序列不仅可以折叠成野生型结构,还可以采样起始结构的结构邻域。
为探究这些相关折叠是否可以在体外得到稳定(即条件序列的pLDDT评分更高),作者为来自TS50的四个目标生成了基于这些采样折叠的序列,包括甘露糖转运蛋白和抗血小板水蛭蛋白。这样采样的序列中只有一小部分以合理的置信度(平均AF2 pLDDT值≥70,与野生型序列相匹配)靠近采样的折叠结构(RMSD≤2埃)。作者还使用Foldseek搜索了PDB、AF2和ESMFold数据库中类似于这些采样折叠的天然蛋白质,但除了一种情况外没有发现任何匹配。这些相邻折叠难以稳定的现象表明,天然蛋白质的折叠空间是稀疏的,跨越或连接这些自然发生的折叠空间的附近结构折叠可能不稳定或仅暂时存在。稍后,作者将研究在自然界中观察到折叠转换或构象灵活性的结构折叠。
生成的蛋白质序列概括了本地序列的生物相关的序列邻域
为了比较Prot_EnT采样的序列空间与生物学采样的序列空间(即我们两个选定目标的天然序列,甘露糖转运蛋白和抗血小板水蛭蛋白),作者基于它们各自的结构采样了10,000个序列。作者还使用MMseqs2提取了与野生型蛋白质相关的天然序列。然后,作者用ESM-1b嵌入对每个序列(采样和MMseqs2)进行嵌入,并将嵌入投影到二维空间。在二维空间中的投影结果揭示了投影空间中蛋白质语言模型视角下的序列相似性。采样序列重现了天然序列的序列空间(图2C)。与具有数千个生物学相关序列的甘露糖转运蛋白序列不同,野生型抗血小板水蛭蛋白序列只有少量相关的天然序列,这些序列稀疏地分布在投影序列空间中(图2C)。相比之下,Prot_EnT采样序列可以填充这个稀疏分布的序列空间(T = 1.0),从而桥接自然界中观察到的序列空间(图2D)。因此,采样序列可以扩展起始结构的局部结构和序列邻域。
生成的序列可以稳定天然序列的结构构象体
接下来,作者提出了两个问题:灵活性是否被编码到结构中,使得作者可以通过序列生成来遍历柔性蛋白质的构象空间?其次,在不同温度下生成的序列的结构是否会反映自然界中观察到的构象状态?为回答这些问题,作者选择了明胶蛋白,一种由severin结构域组成的高度柔性蛋白质。非活性形式表现为封闭构象。加入钙离子会激活一种类似锁闩的机制,导致活性开放构象。
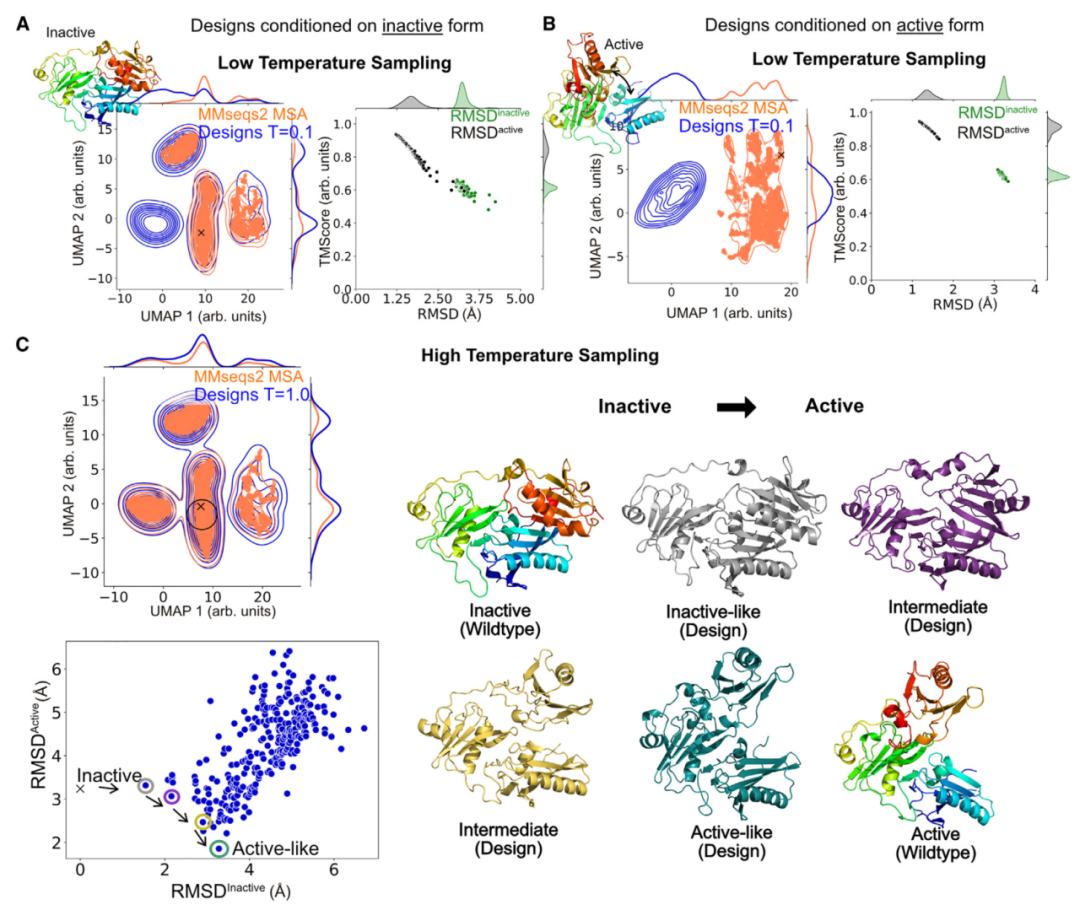
图 3
作者对非活性构象和活性构象进行了条件序列采样(图3A和3B)。在低温(T = 0.1)下,以非活性构象(PDB: 1D0N)为条件的序列重现了通过MMseqs2获得的与天然序列相关的自然序列(图3A,左)。然而,以活性构象(PDB: 1H1V;图3B,左)为条件的序列与自然序列不同。这种差异的一种可能解释是,自然序列被优化为折叠成非活性构象,并且仅在被钙离子触发时才表现出活性构象,而在低温下以活性构象为条件的采样序列对活性构象是最优的,无需激活。在更高温度(T = 0.5)下,为活性构象生成的序列与自然序列重叠(图S6B)。
此外,对于每种构象,作者用AF2折叠了200个随机采样的序列,并计算了结构的RMSD(比较图3A、3B)。以非活性构象为条件的序列显示了广泛的分布,即它们折叠成可能偏离非活性构象的结构,而以活性构象为条件的序列则几乎不偏离活性构象。这一结果表明,非活性结构本质上比活性构象更灵活,这与非活性构象需要预先倾向于构象变化的生物学要求一致。
为了研究采样序列是否能在更高温度下访问替代构象,作者采样了以非活性构象为条件的序列(图3C)。作者用AF2折叠了野生型序列附近的200个序列(用十字标记),并计算了折叠结构与活性和非活性构象的RMSD(图3C,右)。采样序列表现出广泛的构象范围,从接近非活性构象(灰色)到活性构象(青色)以及一些中间构象(黄色和紫色)。
因此,通过在不同温度下采样模型的序列空间并以不同构象为条件,作者可以遍历明胶蛋白的构象空间,从非活性构象到活性构象。
蛋白质界面的残基环境类似于蛋白质内部环境
对于蛋白质-蛋白质界面,作者比较了通用模型、仅界面模型和针对界面微调的通用模型(表1;微调策略I)。仅界面模型PPI_EnT只在来自MaSIF数据集的蛋白质-蛋白质界面数据上训练(无预训练)。微调的Prot_PPI_EnT模型从预训练的Prot_EnT模型开始,随后在MaSIF数据集上进行微调。表1比较了在PPI测试集(PPI300)上使用不同训练方案训练的模型的序列恢复情况。在蛋白质界面上进行微调(Prot_PPI_EnT)相比Prot_EnT模型仅显示出小幅的序列恢复改进,这表明界面残基环境类似于蛋白质核心区域的环境。其他研究也报道了针对单链蛋白训练的模型在蛋白质界面上具有高序列恢复率。
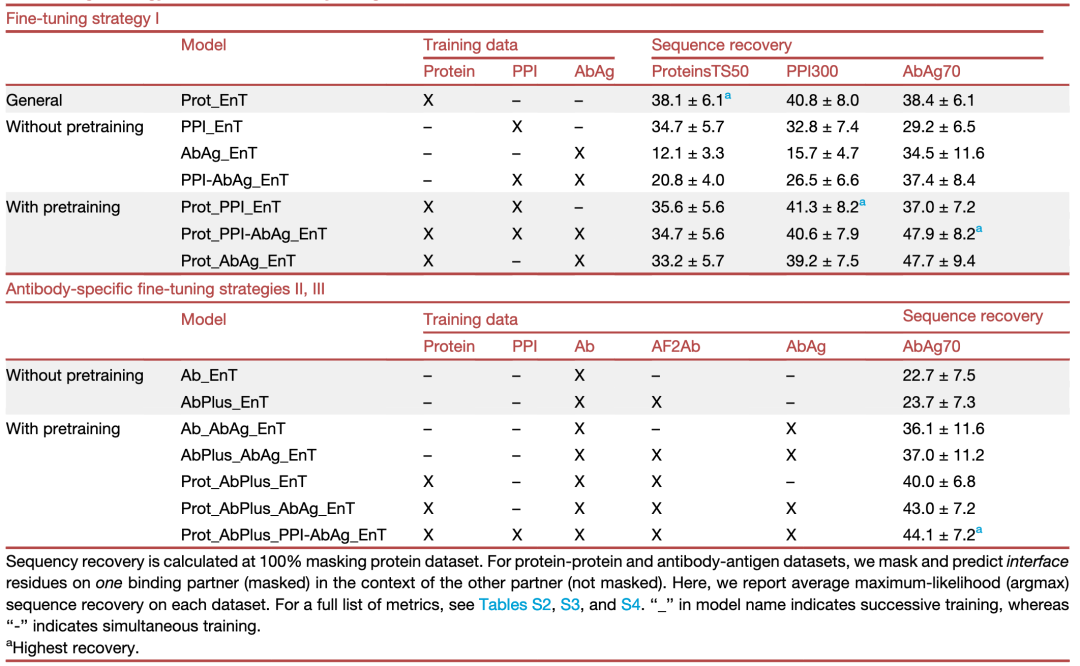
表 1
在数据有限的特定环境中进行训练时,使用仅蛋白质数据集的重要性通过Prot_EnT和Prot_PPI_EnT相比PPI_EnT的优越性能得到了强调。作者还发现,通过从预训练数据集中进行子采样的微调方法能有效防止灾难性遗忘。微调后的Prot_PPI_EnT模型在PPI300测试集上实现了41.9%的序列恢复率,同时在ProteinsTS50测试集上保持了35.3%的序列恢复率。
编译|黄海涛
审稿|王梓旭
参考资料
Mahajan, S. P., Dávila-Hernández, F. A., Ruffolo, J. A., & Gray, J. J. (2025). How well do contextual protein encodings learn structure, function, and evolutionary context?. Cell Systems.
内容中包含的图片若涉及版权问题,请及时与我们联系删除

评论
沙发等你来抢