
DRUGAI
今天为大家介绍的是来自英国InstaDeep公司的一篇论文。探索广阔且大部分尚未被探索的氨基酸序列领域对于理解复杂蛋白质功能和设计新型治疗蛋白质至关重要。虽然生成式机器学习已经推进了蛋白质序列建模,但现有方法在无条件和条件生成方面都不够精通。在本研究中,作者提出贝叶斯流网络(BFNs)——一个最近引入的生成建模框架,可以解决这些挑战。作者提出了ProtBFN,一个在UniProtKB蛋白质序列数据上训练的6.5亿参数模型,它能生成类自然、多样、结构连贯且新颖的蛋白质序列,明显优于领先的自回归和离散扩散模型。此外,作者在来自观测抗体空间的重链上微调ProtBFN,得到了一个特定于抗体的模型AbBFN,用于评估零样本条件生成能力。研究发现,当应用于预测单个框架或互补决定区域时,AbBFN与特定于抗体的BERT风格模型相比具有竞争力或更优。

借鉴词语和氨基酸序列("蛋白质语言")建模之间的相似性,蛋白质自监督模型学会从未标记数据语料库生成新颖序列。例如,BERT风格模型——如进化尺度建模(ESM)系列——被训练用于根据序列其余部分的上下文预测被掩蔽的氨基酸。然而,虽然被训练用于有条件地生成序列的缺失部分,BERT模型并不适合生成全新的蛋白质序列。相反,它们主要用于生成嵌入式表示,用于各种下游任务,如折叠、反向折叠、接触预测和各种其他应用。
随着生成式预训练transformer(GPT)模型在NLP中成为主导范式,等效的自回归蛋白质语言模型——如ProtGPT2、ProtGen和RITA——已被探索用于从头蛋白质序列生成。这些自回归模型从左到右构建序列,每个氨基酸仅以前面的部分序列为条件。由于蛋白质功能取决于三维结构,重要区域通常分布在整个序列中,而不仅仅位于两端。这使得GPT模型在许多实际设计任务中效果较差。实际上,蛋白质序列不同部分之间的复杂和长程相互作用引发了对自回归方法是否是生成蛋白质的最佳选择的怀疑。
鉴于将蛋白质序列严格视为语言结构的局限性,最近的努力已探索了来自其他领域的生成技术。值得注意的是,在图像生成方面表现出色的扩散模型,通过RFDiffusion和AlphaFold3等改编,在结构蛋白质建模中显示出了前景。然而,扩散模型是为连续变量量身定制的,并不自然地转化为离散数据(如氨基酸序列)的建模。虽然所谓的"离散扩散"方法的开发是一个有前途的研究领域——例如,通过在离散空间上定义前向过程,噪声来自有限的扰动集合——但实际上,尚未有任何方法被证明在无条件从头生成和任意条件生成新型蛋白质序列方面都有效。
在此,作者提出最近开发的贝叶斯流网络(BFNs)可以解决上述挑战。BFNs是生成模型,不强制对所有变量的联合分布进行特定分解。此外,BFNs不直接建模数据,而是建模数据分布的连续参数。因此,它们自然地处理非连续数据模态,包括离散变量,同时在概念上与扩散模型保持一致。直观地说,BFNs不是"学习数据",而是"学习关于数据的信念"。由于它们最近才被提出,BFNs尚未在与上述生成方法相同范围的任务中进行测试。
用于蛋白质序列的贝叶斯流网络
生成模型旨在捕获数据集的联合分布p(x),其中每个样本由N个变量组成,表示为x = [x₁, ..., xₙ]。通常,这些模型被训练为从带噪声或损坏的样本中重建原始数据。传统的序列建模技术,如GPT和BERT,通过操作这些变量的子集来创建带噪声的训练目标。具体而言,GPT以自回归方式建模分布,而BERT基于未被遮盖的变量有条件地生成数据。
相比之下,BFNs类似于变分扩散模型,在所有变量上均匀应用噪声。新样本的生成可视为一个连续时间的去噪过程,从随机先验开始,以明确定义的样本结束。与直接建模数据x的传统扩散技术不同,BFNs调整数据分布的参数θ = [θ₁, ..., θₙ],其中p(xᵢ|θᵢ)控制第i个变量的分布。这种区别至关重要,特别是在处理离散数据时,变量是分类正确或错误的。这种中间渐变的缺乏使得在数据上定义平滑的去噪过程(即数据变得单调更准确)成为传统扩散方法应用中的重大挑战。
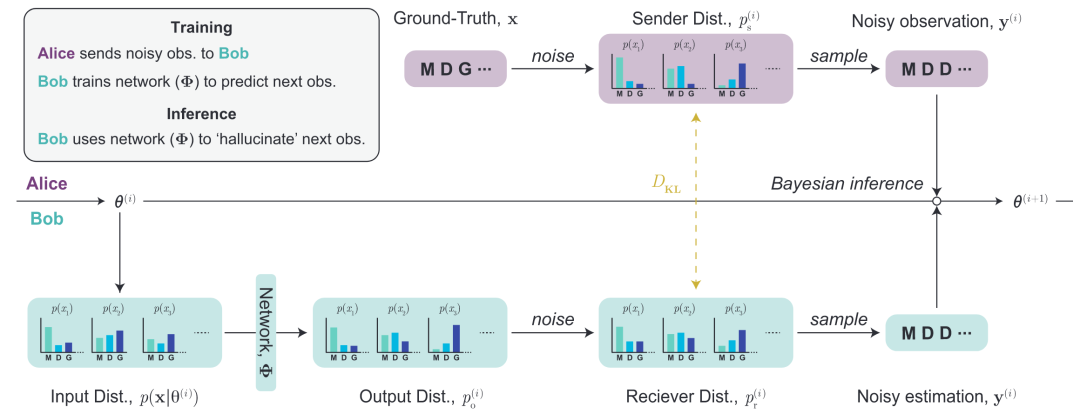
图 1
网络如何训练?用于生成样本的去噪过程可以被视为一种通信协议(见图1),其中Alice通过发送一系列噪声观测值y(1: i) ≡ {y(1), ...y(i)}来向Bob描述真实数据点(例如,一系列氨基酸)。训练BFN等同于Bob学习一个模型,该模型在给定已知噪声的一系列观测的情况下,预测最匹配下一个观测y(i+1)的数据分布。具体而言,应用于蛋白质序列的第i步训练过程如下:
1、Alice从发送者分布(p(i)s)向Bob发送蛋白质(x)的噪声观测(y(i));
2、Bob通过贝叶斯推断,在每个氨基酸的基础上总结所有噪声观测y(1: i),获得输入分布θ(i)的参数;
3、这些参数输入神经网络Φ,预测输出分布(p(i)o)。这一步建模氨基酸的联合分布,改进单变量贝叶斯推断;
4、为获得Bob对下一个噪声观测(y(i+1))形式的信念(belief),输出分布会被加噪,加噪的方式是以与真实数据等效的方式加入噪声,得到接收者分布(p(i+1)r)。
训练目标函数如式1所示,用于最小化N步生成过程中,预测分布与下一个数据样本的真实分布的差别。当N趋近于无穷时,能够推导出一个连续时间损失函数。
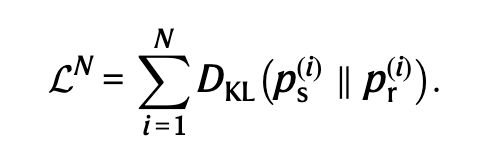
式 1
网络如何推理?当从模型无条件地采样完整蛋白质序列时,没有预设的真实数据。因此,Bob需要幻想出一个Alice,前一步的接收者分布扮演发送者分布的角色(p(i)s ≈ p(i-1)r)。重复此过程N次可从学习的分布中生成样本。虽然条件生成的简单方法是对具有真实数据的变量使用发送者分布,而在其他地方使用接收者分布,但作者发现这种方法不会收敛到真实的条件分布。相反,作者将此方法与时序蒙特卡洛(sequential Monte Carlo,SMC)采样相结合;这确保了在学习的联合分布下,采样变量与固定变量保持一致。
使用上述方法,作者训练了一个650M参数的模型ProtBFN,训练数据集是一个涵盖已知蛋白质空间的精选序列数据集。该数据集考虑了UniProtKB中的非假设性蛋白质序列,作者将这个经过精选和聚类的数据集称为UniProtCC。在评估ProtBFN的能力时,作者将其与领先的蛋白质序列生成自回归模型和离散扩散模型进行了比较——分别是ProtGPT2和EvoDiff。需要注意的是,ProtGPT2和EvoDiff都是在UniRef50数据集上训练的,而与UniProtCC不同,UniRef50包含了假设性或未知存在的蛋白质。
通过仅在UniRef50中里的高置信度序列上训练ProtBFN,作者确保它专注于生物学相关的可能蛋白质流形。作者的评估分析了每个模型生成的10,000个蛋白质序列,并将这些序列与从UniProtCC和UniRef50数据集中随机选择的10,000个序列子集进行了比较。
ProtBFN模型性能
ProtBFN的主要目标是学习自然蛋白质的分布。尽管没有单一指标可以明确评估生成的蛋白质样本的自然性,但可以计算各种统计和生物物理特性并与预期的自然分布进行比较。为此,图2展示了一系列此类实验,从中作者可以推断出ProtBFN不仅与它训练的自然分布相匹配,而且比在各自训练数据集上的自回归和离散扩散基准模型更忠实地匹配。
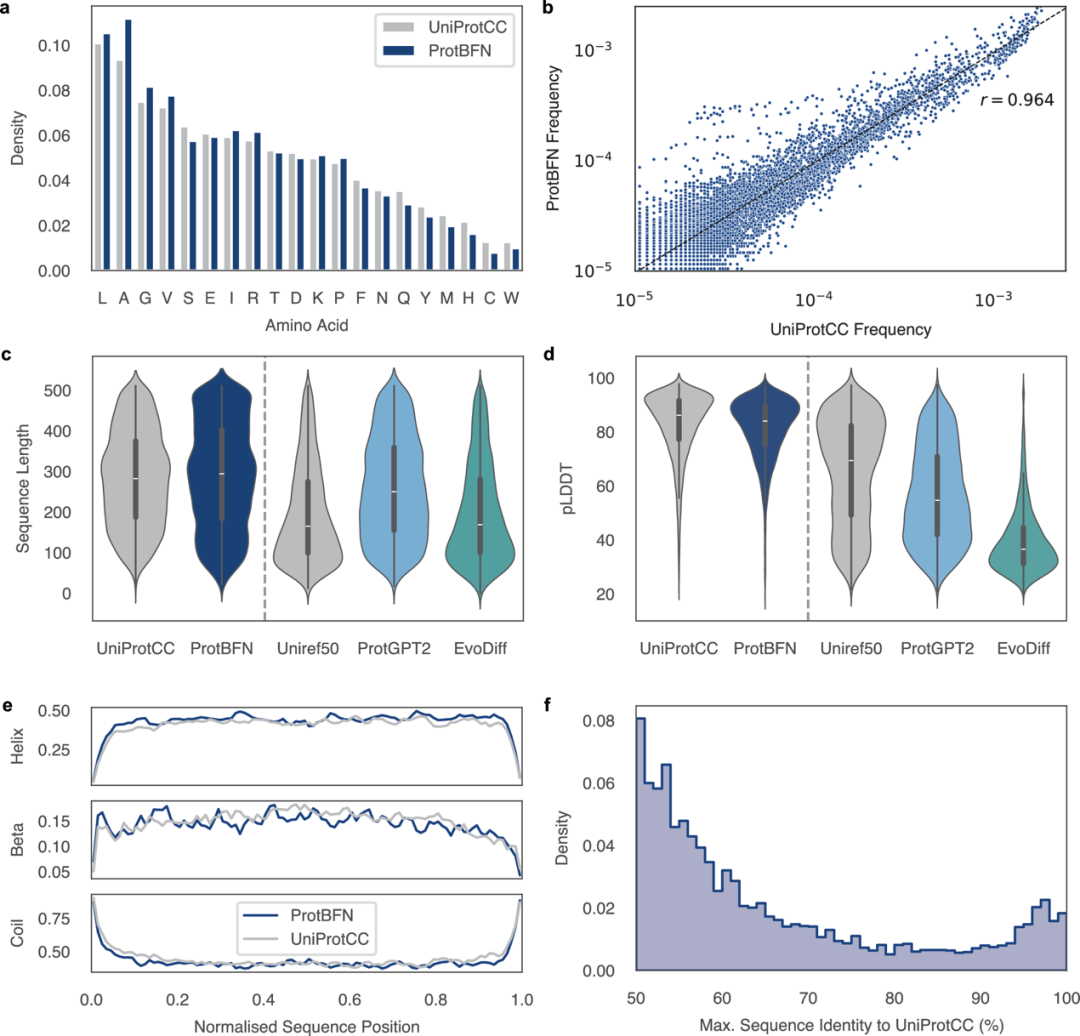
图 2
检验ProtBFN生成的蛋白质序列集合可行性的直接测试是检查氨基酸和低聚物(oligomers)出现的频率,因为这些与遗传密码的稳健性密切相关。这些测量(图2a、b)与UniProtCC分布高度一致。然而,虽然这些频率指标具有指示性,但它们本身并不能确认单个样本的连贯性,因此,作者还计算了每个样本的指标。ProtBFN生成的序列具有类似自然的长度分布(图2c),这是作者建模底层分布能力的另一个指标。相比之下,ProtGPT2在这一指标下明显偏离了UniRef50分布,而EvoDiff人为地匹配了目标分布,因为序列长度是在生成前预先选定的。
为了进行结构分析,作者首先利用NetSurfP-3.0(一种二级结构和相对溶剂可及性预测方法)来为序列中的每个残基添加结构信息注释。作者再次发现ProtBFN在这些指标上匹配自然分布,即使考虑这些属性如何随序列中的位置变化而变化(图2e)。
为了评估整体结构特性,作者使用ESMFold的平均预测局部距离差异测试(pLDDT)作为对预测结构置信度的度量(图2d)。蛋白质序列中相距较远的残基之间的相互作用对确定整体结构至关重要。因此,更高的pLDDT分数表明ESMFold可以识别与其训练数据中发现的相似的相互作用。实际上,作者观察到ProtBFN持续产生能获得高pLDDT分数的序列,与自然发生的蛋白质的分数紧密匹配。ProtGPT2和EvoDiff偏离其训练分布的情况证明了该指标对蛋白质全局连贯性的敏感性,即使考虑到UniRef中较低得分蛋白质数量比UniProtCC多的因素。
最后,为了确认ProtBFN是在生成新颖的蛋白质而不是记忆训练数据,作者在UniProtCC训练数据中搜索每个生成序列的最近匹配(图2f)。结果表明,生成的序列很可能是新颖的,其中4444个(分别有8851个和9489个)生成的样本与最近匹配的序列同一性小于50%(分别为80%和95%)。
ProtBFN有效覆盖已知蛋白质组
蛋白质组的广泛覆盖确保模型已经学习了蛋白质序列的多样性,因此可用于开发各种功能性蛋白质。在确认了ProtBFN生成序列的自然性和新颖性后,作者接下来评估这些序列提供的蛋白质组覆盖范围。为此,作者首先使用mmseqs2将生成的序列与UniRef50进行比对(表1),因为UniRef50代表了涵盖已知蛋白质多样性的非冗余蛋白质聚类集。
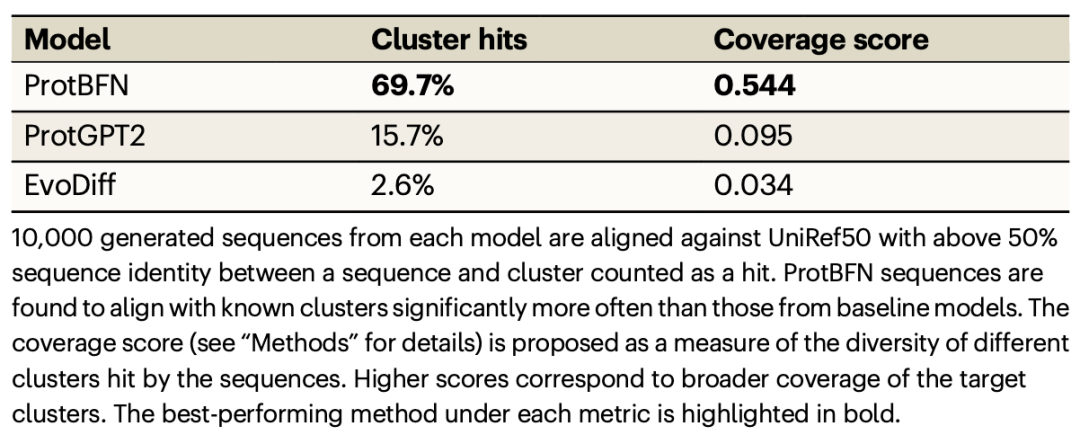
表 1
发现ProtBFN的69.7%的序列与已知的UniRef50聚类对齐(≥50%序列同一性)。这一比例显著高于ProtGPT2和EvoDiff,尽管这些模型均匀地在UniRef50聚类上进行训练。由于采样序列数量(10,000)远小于UniRef50聚类数量(约6500万),作者将覆盖率测量为如果从模型的训练分布中抽取10,000个样本,观察到的与预期的独特聚类命中率之比。结果表明,ProtBFN比基准方法提供了更好的蛋白质空间覆盖率。这一结果与图2f相结合,强化了ProtBFN创建了新颖序列的观点,这些序列仍然覆盖了UniProtKB中存在的功能性蛋白质空间。
为了可视化分布覆盖,作者使用最先进的蛋白质语言模型计算蛋白质序列的嵌入,并将其投影到二维空间(图3)。ProtBFN和UniProtCC可视化分布之间的紧密匹配突显了生成的序列在这个生物学意义的表示空间中提供了对自然分布的广泛覆盖。
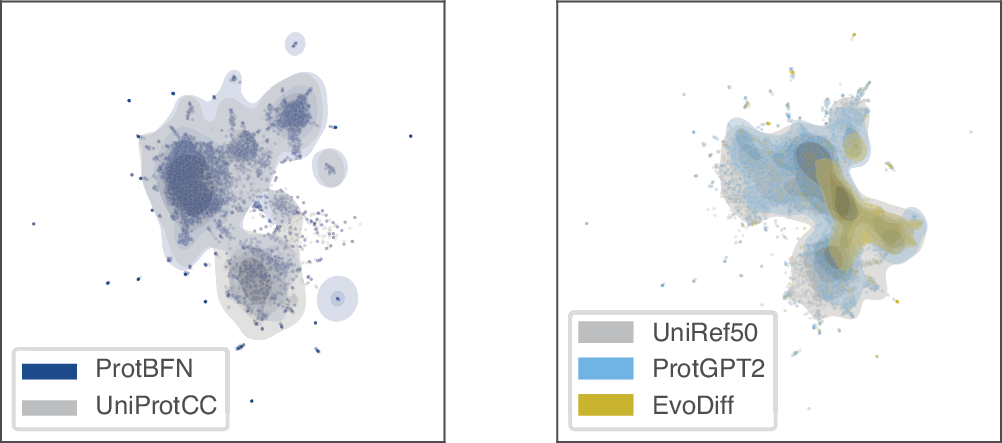
图 3
ProtBFN生成具有新颖序列的球状结构模块
蛋白质功能与结构紧密相连。因此,为了表征可能与自然界观察到的结构有实质性差异的全新蛋白质序列,作者分析了它们与实验确定的蛋白质构象的对应关系。CATH S40数据库包含约30,000个非冗余的、实验性解析的蛋白质结构域,提供了已知结构多样性的广泛覆盖。作者使用模板建模(TM)分数比较了从每个模型采样的2000个序列与CATH S40结构域的结构相似性。TM1和TM2分数分别针对生成蛋白质的长度和CATH S40结构域的长度进行标准化。一般认为,TM分数高于0.5表示具有相同折叠。为了避免仅将CATH结构域的片段与生成的蛋白质匹配,或相反,正匹配要求TM1和TM2分数均超过0.5。
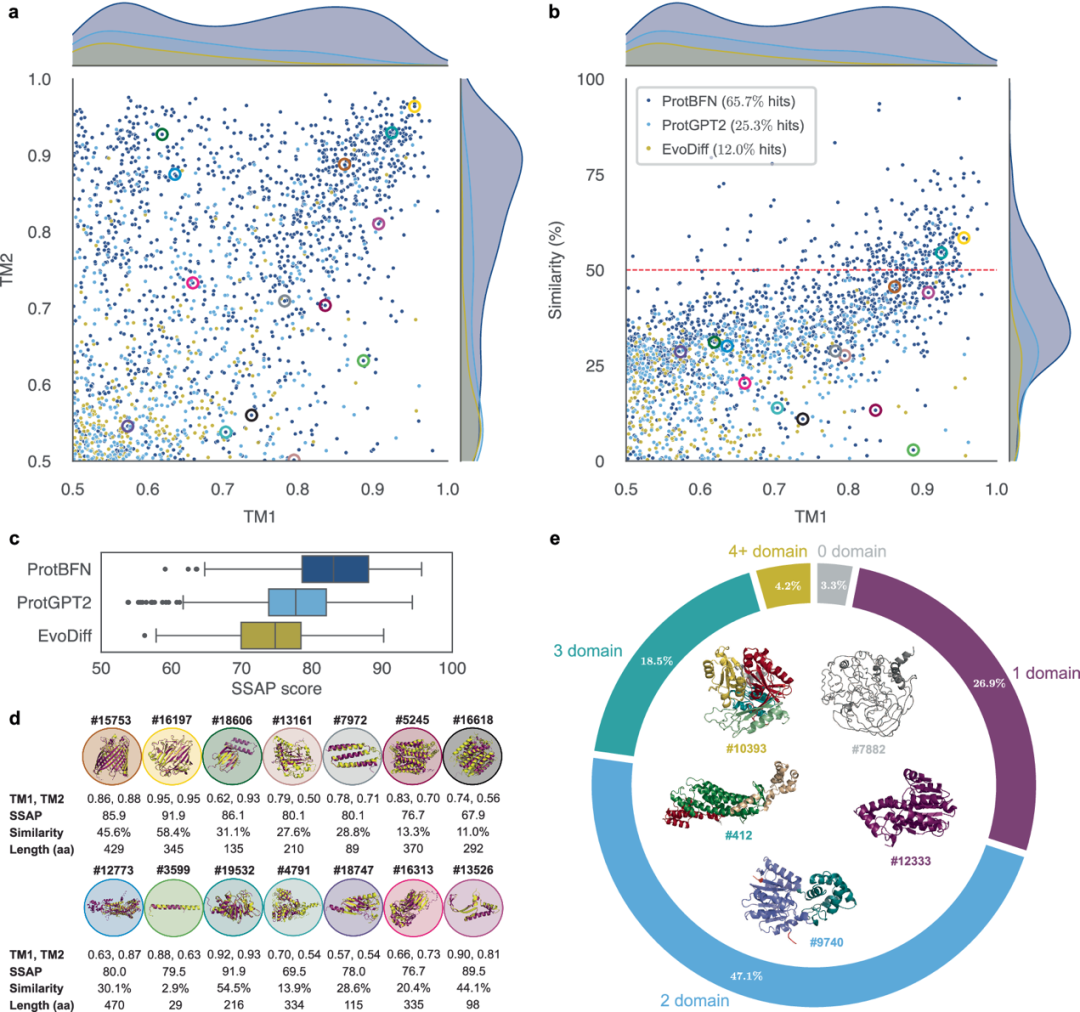
图 4
ProtBFN达到了65.7%的CATH命中率,超过了ProtGPT2(25.3%)和EvoDiff(12.0%)。此外,这些命中质量更高,ProtBFN命中的68.0%具有超过80的序列结构对齐程序(SSAP)分数。这对应于CATH分类中的最高相似性级别(同源超家族),而ProtGPT2和EvoDiff分别为38.0%和19.2%(图4c)。此外,接近完全命中的比例,即两个TM分数接近1(图4a),表明ProtBFN更频繁地生成能折叠成已知结构域的较长序列。这一点得到了ProtBFN样本在所有采样蛋白质长度上保持高SSAP分数的事实支持。
然而,尽管有高度的结构对应性,ProtBFN的序列与其CATH S40目标的序列相似性较低,80.4%的命中具有低于50%的序列相似性(图4b)。这种能够产生具有新颖序列的可识别球状折叠的能力是将ProtBFN用于理性蛋白质设计的先决条件,并表明它有意义的能力超越了训练数据并探索未知的蛋白质组。
再次考虑生成样本的多样性很重要,如图4d所示,ProtBFN在生成功能复杂的蛋白质结构域的结构多样性数组方面也表现出色。该模型涵盖了CATH目录中记录的广泛类别,包括α-螺旋(样本7972、5245、16618、12773、3599)、β-折叠片(15753、16197、18606)、α-β混合(19532、4791、18747、16313)和不规则结构域(13525)。ProtBFN还有效地模拟了各种功能类型,如跨膜蛋白,包括孔蛋白(15753)和转运蛋白(16618),以及酶(2773、13161)。此外,生成的球状蛋白跨越了小型(18606、7972、18747)和大型(16197、5245、4791)结构域,不受CATH类别限制。
最后,作者使用Merizo(一种结构域分割工具)来理解ProtBFN样本的结构域分布(图4e)。接近一半的测试样本有两个结构域,26.9%有单一结构域,22.7%有三个或更多结构域。有趣的是,多结构域构造通常表现出结构域-结构域界面,以不同类型的相互作用为特征,而不仅仅是通过无序环连接。这突显了ProtBFN能够生成具有全局连贯相互作用的蛋白质,即使相互作用的残基在序列空间中是分离的,并且属于不同的折叠。更广泛地说,这一分析展示了ProtBFN学习分布的结构和功能深度,突显了BFN生成在多种生物学和生物技术应用中的潜在适用性。
编译|黄海涛
审稿|王梓旭
参考资料
Atkinson, T., Barrett, T.D., Cameron, S. et al. Protein sequence modelling with Bayesian flow networks. Nat Commun 16, 3197 (2025). https://doi.org/10.1038/s41467-025-58250-2
内容中包含的图片若涉及版权问题,请及时与我们联系删除

评论
沙发等你来抢