
DRUGAI
今天为大家介绍的是来自德国慕尼黑大学Christian L. Müller率领的团队发表的一篇论文。抗生素耐药性正在全球范围内日益严重,这使得常见抗生素的效力不断下降。通过实验方法测试新化学化合物的抗菌活性不仅耗时,而且成本高昂。尽管以化合物为中心的深度学习模型有望加速这一筛选过程,但现有方法通常需要大量专门的训练数据。本研究提出了一种轻量级计算策略用于抗菌素发现,该策略基于MolE(Molecular representation through redundancy reduced Embedding)。MolE是一种自我学习的深度学习框架,它能够利用未标记的化学结构数据自动学习分子的通用特征表示。作者将MolE的表示学习能力与已有的实验验证数据相结合,开发出一个通用预测模型,可以评估化合物的抗菌潜力。作者的模型成功识别了近期发现的生长抑制化合物,这些化合物在结构上与现有抗生素有明显区别。更重要的是,通过这一方法,作者发现并实验证实了三种原本用于人类治疗的药物可以有效抑制金黄色葡萄球菌的生长。这一研究框架为加速抗生素发现提供了一种可行且经济高效的策略,有望帮助解决日益严峻的抗生素耐药性挑战。

面对日益严重的抗生素耐药性问题,开发新型抗生素已成为当务之急。传统上,科研人员通过筛选大型化学库来寻找潜在的抗生素候选物,但这种方法成功率仅有1-3%,且耗费大量人力物力。更令人担忧的是,现有化学库中分子多样性有限,难以验证新发现或新合成的化学物质。在这一背景下,研究团队开发了一种创新的深度学习辅助策略,可以大幅提高筛选效率。这一方法采用两阶段深度学习架构(如图1所示):
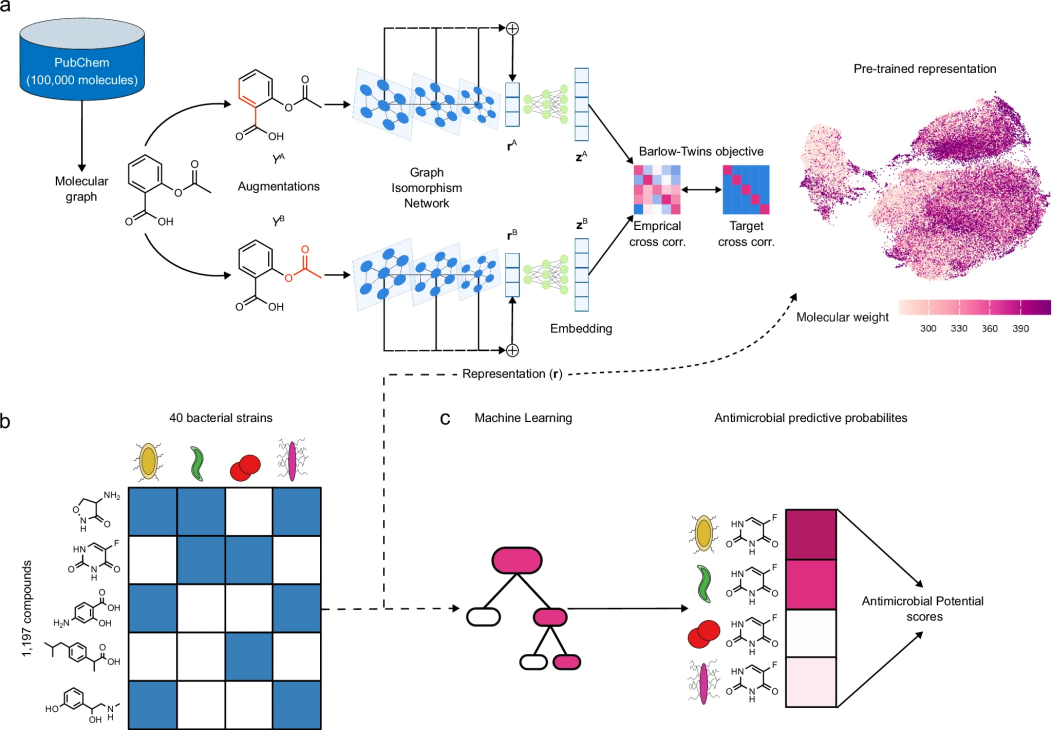
图 1
第一阶段使用名为MolE的自监督学习框架(分子冗余减少嵌入表示法)进行分子特征提取。MolE能够从PubChem数据库中大量未标记的化学结构中学习通用的分子表示。第二阶段利用MolE学到的分子表示,结合公开可获取的FDA批准药物对多种细菌的抑制效果数据,计算出一系列抗菌潜力(AP)评分,用于化合物筛选优先级排序。
与以往需要大量特定任务训练数据的方法不同,MolE采用图同构网络(GINs)进行表示学习,并创新性地将Barlow-Twins非对比学习框架应用到分子领域。这使得该方法能够在多种分子性质预测任务上取得竞争性能。研究证明,MolE计算的抗菌潜力评分能够准确反映不同结构化合物(如Halicin和Abaucin)的广谱和窄谱抗菌活性。在一个包含2000多种化合物的库中,研究团队成功识别出约200种具有高抗菌潜力评分的化合物。更令人鼓舞的是,团队从预测的高抗菌潜力但无已知抗菌活性的分子中,优先选择了六种进行实验验证,结果证实其中三种对人类病原体金黄色葡萄球菌具有显著抑制作用,成功率远高于行业平均水平。这一研究为微生物学家提供了一种通用且经济高效的方法,用于优先筛选和发现具有抗生素特性的新分子,有望加速新型抗生素的开发进程,应对全球抗生素耐药性危机。
MolE:一种智能分子表示学习方法
开发新型抗生素的关键前提是能够高效、通用地表示分子结构。研究团队开发的MolE(分子冗余减少嵌入表示法)框架正是为此而设计。这一创新方法采用自我学习机制,无需人工标注数据,就能自动理解分子的结构特征。MolE的工作原理类似于人类学习识别物体:通过反复观察不同角度的同一物体来理解其本质特征。具体来说,系统首先将分子的文本表示(SMILES)转换为图结构,其中原子作为节点,化学键作为边。然后,系统通过“遮盖”分子结构的部分内容,创建同一分子的两种变体,并学习识别它们代表相同分子的能力。这一过程使MolE能够专注于分子的本质特征,而非表面细节。
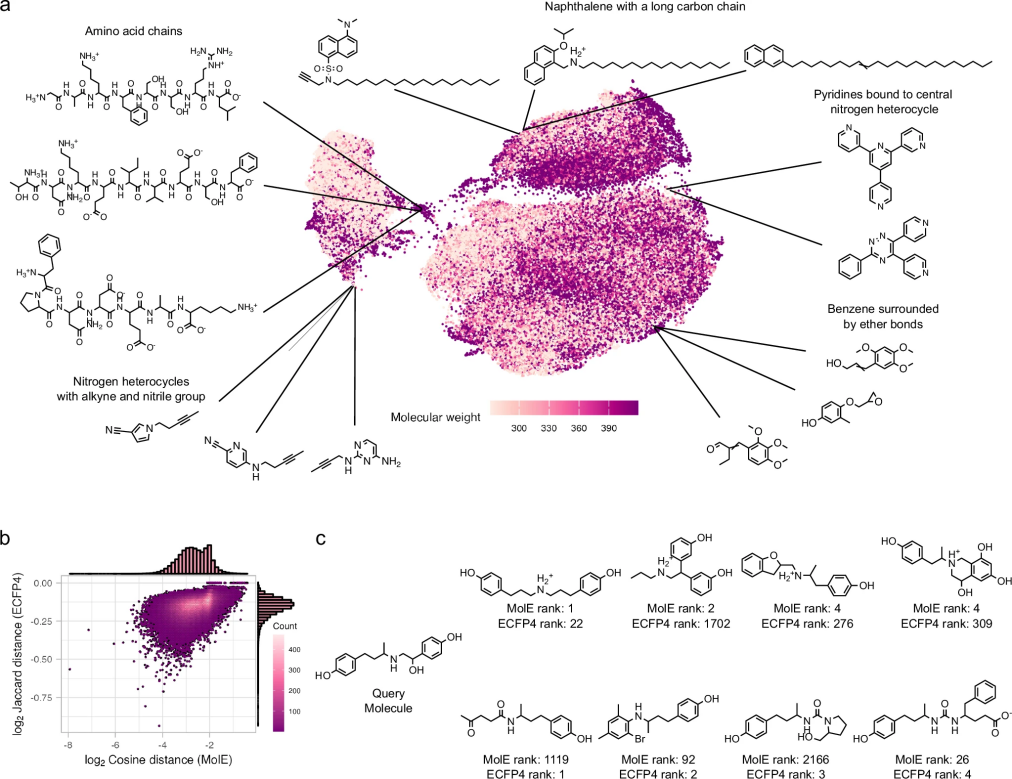
图 2
研究表明,MolE能够智能地识别分子的关键结构特征。如图2所示,具有相似功能基团或拓扑结构的分子在MolE的表示空间中被放置在一起。例如,含有萘基团连接长碳链的化合物、含有吡啶环的化合物、含有醚键的苯环化合物,以及含氮杂环化合物等都被分别聚类。特别值得注意的是,MolE甚至能够识别短氨基酸链的相似性,这些肽类分子在表示空间中形成了独特的聚类。与传统的分子指纹表示法(ECFP4)相比,MolE能够捕捉更多化学上有意义的特征。以雷克托胺(Ractopamine)为例,MolE识别出的最相似分子都共享两个酚基团和一个胺基团,而ECFP4只能识别一个酚环和一个甲基。这表明MolE能够更全面地理解分子的化学结构。
在分子性质预测方面,MolE结合机器学习算法(如XGBoost)在多项任务中表现出色。在六项分类任务中,MolE在四项任务上取得最佳结果,平均性能提升3%。特别是在临床毒性预测(ClinTox)基准测试中,MolE达到了92.85%的准确率,甚至超过了一些需要数百万训练样本的大型模型。最令人印象深刻的是,MolE只需约10万个未标记分子结构就能学习到有意义的表示,计算资源需求远低于其他需要数百万样本的方法。这种高效性使MolE特别适合那些标记数据稀缺的任务,为抗生素发现提供了一种经济实用的计算工具。
MOIE精准预测化合物抗菌活性
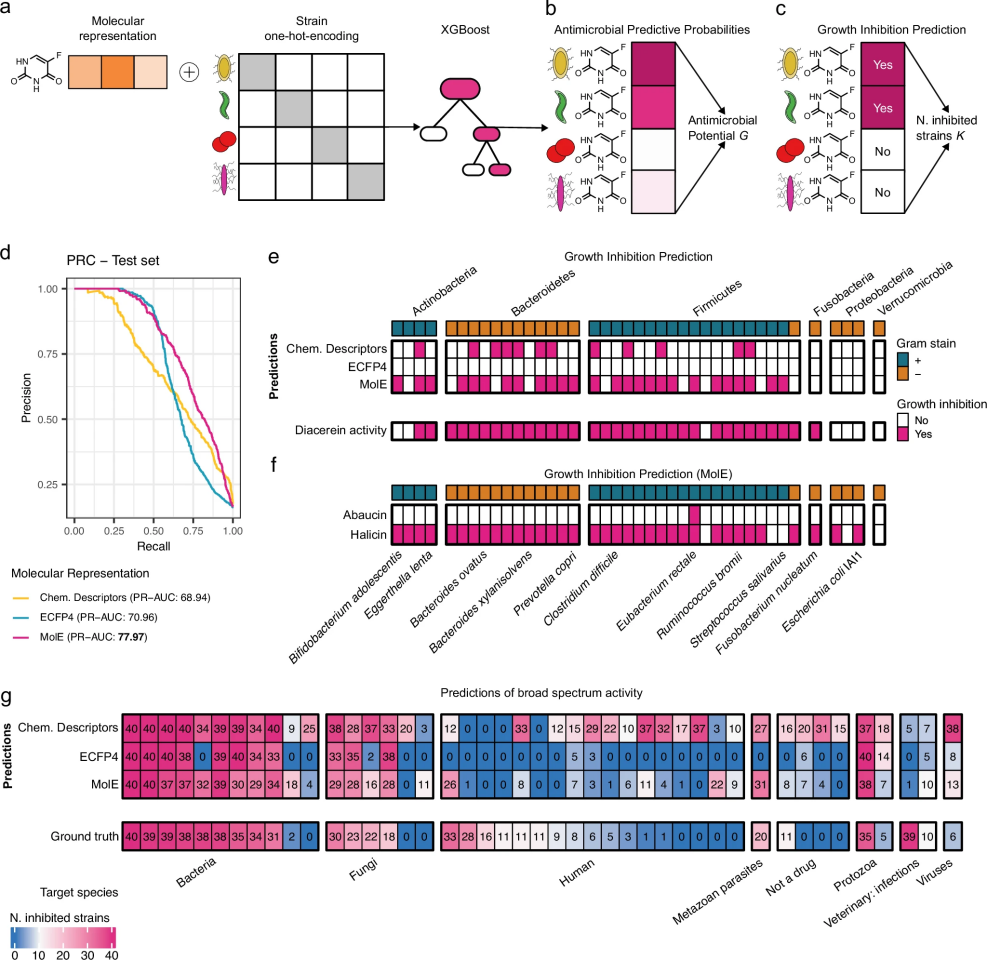
图 3
在MolE框架的第二阶段,研究团队将其强大的分子表示能力应用于抗菌药物筛选领域。如图3所示,他们利用一个包含1197种市售药物对40种人类肠道细菌影响的公开数据集,训练了名为MolE-XGBoost的预测模型。这一模型能够为任何化合物生成一个“抗菌活性指纹”——预测该化合物对40种不同细菌的抑制概率。这种预测能力有两个关键应用:一是判断化合物是否能抑制特定细菌的生长;二是评估化合物的抗菌谱范围——即它能抑制多少种不同的细菌。所谓“广谱”抗生素能够对抗多种细菌,而“窄谱”抗生素则只针对特定类型的细菌有效。
与现有方法相比,MolE-XGBoost展现出明显优势。在精确度和召回率(两个衡量预测准确性的重要指标)方面,它大幅超越了传统的ECFP4指纹方法和其他使用化学描述符的模型。一个典型例子是对人类靶向药物Diacerein(洛芬待因)的预测——MolE-XGBoost正确识别了该药物在实验中显示的33种抑制效果中的25种,而基于ECFP4的模型完全未能发现其任何抗菌活性。更令人印象深刻的是,MolE-XGBoost能够“重新发现”近期突破性研究中发现的新型抗生素。它准确预测了Halicin(一种由人工智能发现的广谱抗生素)对大肠杆菌和艰难梭菌的抑制作用。对于Abaucin(一种窄谱抗菌药物),模型也正确识别了其高度特异性的活性模式。
在对24种已知具有广谱抗菌活性的化合物的测试中,MolE-XGBoost展现出最高的预测准确率。而传统的ECFP4模型未能识别大多数人类靶向药物的抗菌活性,基于化学描述符的模型则倾向于过度预测,导致大量假阳性结果。特别值得一提的是,MolE-XGBoost成功识别了5种具有广谱活性的化合物,这些化合物是ECFP4模型完全忽略的。这些结果表明,MolE-XGBoost不仅能准确预测已知抗菌药物的活性,还能发现那些原本被开发用于其他治疗目的但具有潜在抗菌特性的药物。这种能力对于药物重新定位和新型抗生素发现具有重要意义,为应对日益严重的抗生素耐药性危机提供了新的希望。
MOIE预测化学库中的抗菌潜力
研究团队使用MolE-XGBoost模型预测了一个包含2,320种FDA批准药物、食品同源产品和人体内源代谢物的正交化学库中的抗菌潜力评分。他们设计了四种抗菌潜力(AP)评分变体来对个别化合物进行排序:(i)预测被抑制的菌株总数K;(ii)所有40个菌株估计概率的log2几何平均值G;(iii)所有22个革兰氏阳性菌株概率的log2几何平均值G+;(iv)所有18个革兰氏阴性菌株概率的log2几何平均值G-。
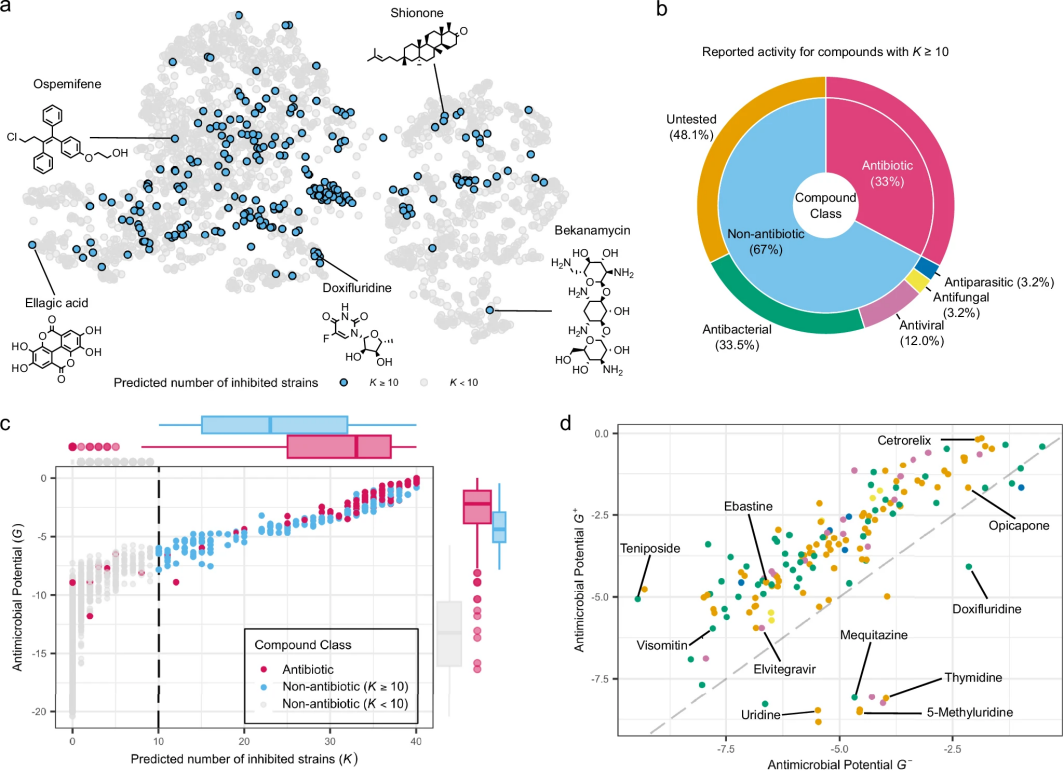
图 4
如图4a所示,在MolE基础的UMAP嵌入中,2320种化合物中有235种被预测为广谱抑制剂化合物。在这些化合物中,158种是非抗生素。文献回顾显示,这158种化合物中有53种先前已被报道抑制各种细菌物种的生长,如天然产物鞣花酸和紫杉酮,以及人类靶向药物氟尿嘧啶。这33%的命中率大大改善了大规模化学筛选常见的1-3%的命中率。
研究团队进一步评估了所提出的AP评分的有效性,测试了它们对化学库中93种已知抗生素的排序能力。图4c显示了所有化合物的一般AP评分G与预测抑制菌株数量K的分布。观察到强大的排序能力,在大AP评分处抗生素化合物富集。此外,93种抗生素中有77种被预测抑制10种或更多菌株,证实两种评分方案对未见过的抗生素化合物都有很好的泛化能力。
最后,研究团队通过同时对先前预测为广谱的158种非抗生素化合物进行排序,评估了针对革兰氏阳性和革兰氏阴性菌株的精细AP评分的区分潜力。与当前关于革兰氏阳性细菌对化学应激源敏感性的知识一致,大多数化合物对革兰氏阳性菌株的AP评分高于对革兰氏阴性菌株的评分。然而,观察到核苷酸类似物,如尿苷和尿苷衍生物,被预测对革兰氏阴性菌株更活跃,证实了最近的证据,即尿苷分子是氨基糖苷类抗生素对抗大肠杆菌的有力辅助剂。
AI预测与实验验证
为了验证MolE-XGBoost模型预测的可靠性,研究团队进一步探索了其抗菌潜力评分与实际实验数据之间的关系。他们通过广泛文献调研,收集了158种被预测具有广谱抗菌活性的非抗生素化合物的实验数据。在这些研究中,科学家们使用“最小抑菌浓度”(MIC)来衡量化合物的抗菌效力——这一指标表示能够抑制细菌生长所需的最低药物浓度,数值越低意味着抗菌效力越强。研究团队发现,31种化合物在实验中显示出较强的抗菌活性,其MIC值不超过128微克/毫升。
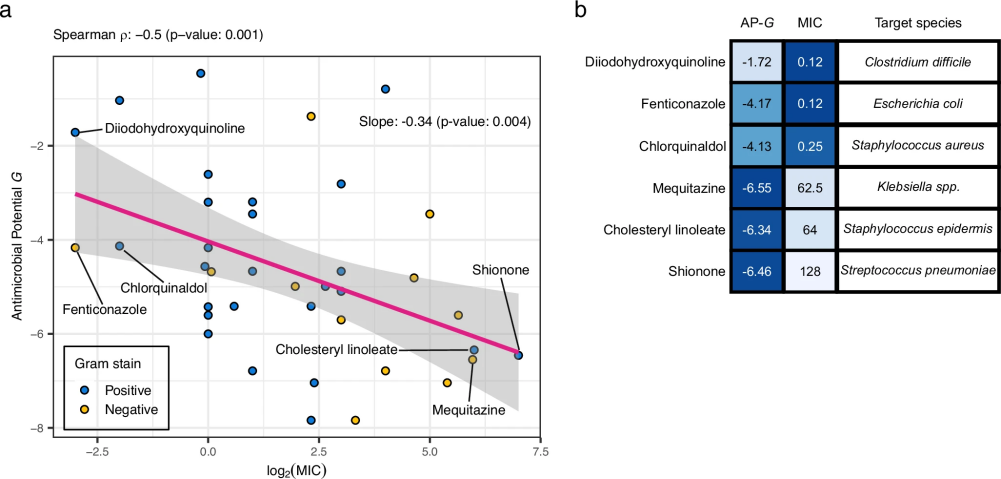
图 5
分析结果令人信服:如图5a所示,MolE-XGBoost预测的抗菌潜力评分与实验测定的最小抑菌浓度之间存在显著的负相关关系(相关系数为-0.5)。这意味着模型给予高评分的化合物,在实验中确实表现出更强的抗菌能力(需要更低的浓度就能抑制细菌生长)。这一关系对革兰氏阳性细菌(蓝色标记)和革兰氏阴性细菌(黄色标记)均成立。
图5b进一步展示了抗菌效力最强和最弱的几种化合物,以及它们能抑制的细菌种类。值得注意的是,这些实验数据中包含了一些模型训练时从未见过的细菌种类,如肺炎链球菌、金黄色葡萄球菌和克雷伯菌等。模型对这些“陌生”细菌的准确预测,证明了其良好的泛化能力——能够应用于新的化合物和细菌组合。研究团队还发现,针对特定类型细菌的抗菌潜力评分(G+和G-)与相应细菌类型的最小抑菌浓度也存在类似的相关性。这进一步证实了评分系统的准确性和实用性。对比之下,当使用传统的ECFP4分子表示方法时,预测评分与实际抗菌效力之间没有显著关联。此外,ECFP4方法识别的高评分化合物中,能在实验中证实有效(MIC ≤128μg/mL)的数量也明显少于MolE-XGBoost方法。
化合物生成验证
研究团队在MolE-XGBoost AP评分的指导下,从发现库中选择了六种化合物进行实验验证:Cetrorelix、Ebastine、Elvitegravir、Opicapone、Thymidine和Visomitin。选择这些化合物的标准是:(i)预测为广谱(K≥10);(ii)AP评分G+ > −10和G− > −10;(iii)覆盖各种功能和化学结构;(iv)商业可获得。重要的是,所选化合物在结构上都与训练集中的抗生素不同(Tanimoto相似性≤0.3)。除Visomitin外,这些化合物此前未被报道抑制细菌或真菌菌株的生长。

图 6
如图6所示,六种测试化合物中有三种被证实对革兰氏阳性病原体金黄色葡萄球菌的生长有可测量的影响。最强的效果观察到的是Elvitegravir,它在8μg/mL的浓度下抑制了金黄色葡萄球菌的生长。此外,Opicapone在128μg/mL的浓度下显著限制了金黄色葡萄球菌的生长,最大光密度为0.38±0.01。在较低的16μg/mL浓度下,它延长了滞后期的持续时间和种群倍增时间约1小时。最后,Ebastine将金黄色葡萄球菌的滞后期延长了约2小时。值得注意的是,实验重新发现了Visomitin的广谱活性,表明它可以在64μg/mL的浓度下抑制铜绿假单胞菌和肺炎克雷伯菌的生长,从而扩大了已知对这种化合物敏感的物种列表。
虽然研究团队没有观察到对Cetrorelix和Thymidine的生长抑制反应,但它们预测的抗菌潜力可以通过训练集中存在的影响细菌生长的类似分子来解释,如叠氮胸苷。这种药物是Thymidine的化学类似物,先前已被证明抑制十二种菌株(九种革兰氏阴性和三种革兰氏阳性)的生长。因此,模型预测Thymidine对革兰氏阴性菌株特别有效。
讨论
面对抗菌药物发现中的数据稀缺问题,MolE框架创新性地利用大量未标记分子结构进行深度学习,捕捉关键化学特征,显著提升了分子性质预测的准确性。基于MolE的预训练表示,作者的XGBoost模型能准确评估化合物的抗菌潜力,成功重新发现了如Halicin等新型抗生素,并识别了传统方法容易忽略的广谱抗菌物质。作者设计的抗菌潜力评分系统可靠地预测了化合物对不同细菌的抑制能力,在实验验证中取得了显著成果——六种测试化合物中有三种对金黄色葡萄球菌显示出明显抑制效果。这些化合物结构上与已知抗生素截然不同,证明了系统发现创新抗菌物质的能力。随着耐药性威胁不断增加,这种计算引导的药物发现方法为加速新型抗生素研发提供了高效途径。
编译|于洲
审稿|王梓旭
参考资料
Olayo-Alarcon R, Amstalden M K, Zannoni A, et al. Pre-trained molecular representations enable antimicrobial discovery[J]. Nature Communications, 2025, 16(1): 3420.
内容中包含的图片若涉及版权问题,请及时与我们联系删除

评论
沙发等你来抢