
DRUGAI
理解和操控分子在不同溶剂环境中的动态行为,是药物发现和有机合成领域中的重要研究方向。分子动力学(MD)模拟通过显式地加入溶剂分子,被认为是在给定力场精度下计算这类构象集合的“金标准”,它能够补充实验发现并支持对实验结果的解释。然而,传统方法往往面临计算成本高(显式溶剂)或精度不高(隐式溶剂)的问题。该研究展示了如何利用基于图神经网络(GNN)的隐式溶剂方法(GNNIS)快速计算小分子在39种常见有机溶剂中的构象集合,并高精度地重现显式溶剂模拟结果。该研究通过核磁共振(NMR)实验对该方法进行了验证,从而识别出对实验可观测量贡献最大的构象。该方法将准确预测构象集合所需的时间从几天缩短至几分钟,同时预测结果与实验值的误差控制在一个k BT以内。
目前上市的大多数药物都是低分子量的有机化合物,通常被称为“小分子”。在溶液中,柔性的分子处于动态状态,会形成一个遵循玻尔兹曼分布的不同构象的集合,这些构象分布依赖于所处溶剂的性质。不同溶剂之间的弱相互作用可能差异极大,从而导致相应构象集合的显著差异。因此,即使是微小的构象群体变化,对于许多应用场景也是至关重要的。这些应用包括区域选择性和立体选择性分子的合成,膜渗透性的优化与理解,以及分子物理性质的精细调控。
此外,不同的构象集合还会导致不同的实验观测结果,从而在很大程度上影响实验技术的解释。例如,核磁共振(NMR)、红外光谱(IR)以及振动圆二色谱(VCD)等实验技术的观测结果均受到构象的影响。虽然带有显式溶剂分子的分子动力学(MD)模拟(如有需要可配合增强采样技术)是研究小分子在溶液中构象分布的有力工具,但其本质上受限于较高的计算成本。在这类模拟中,目标分子(溶质)被置于一个充满溶剂分子的模拟盒中,整个系统在时间维度上进行推进。这种方法效率低下,因为构象的概率是通过模拟过程中重复出现的次数估计的,而不是直接从其自由能出发获得的,而自由能在显式溶剂模拟中并不容易获取。
尽管如此,显式溶剂模拟目前仍被视为“金标准”,因为隐式溶剂模型(如泊松-玻尔兹曼(PB)模型、快速解析连续溶剂处理(FACTS)、广义Born(GB)模型等)尚未达到理想的精度。为填补这一空白,已有研究提出使用机器学习方法。此前的研究表明,针对水这一溶剂,基于图神经网络(GNN)的模型在训练时使用来自显式溶剂模拟的参考力数据,能够以概率方式表达溶剂效应,从而以显著减少的自由度准确地模拟体系。这大大降低了计算负担,同时获得了与显式溶剂模拟相当的结果。该方法受到Δ-learning思想的启发,并在模型中引入传统的GB-SA模型的函数形式作为物理正则项,以确保模型符合正确的物理行为。
本研究基于GNN的隐式溶剂模型(GNNIS)推广至39种常见的有机溶剂,并展示该方法如何快速识别小分子在这些溶剂中玻尔兹曼加权的构象集合。为实现不同溶剂的编码,本研究使用一种嵌入方案,使得模型能够自行学习如何表达每一种溶剂。这一方法可以将一组随机生成的初始构象(例如来自构象生成器)优化为玻尔兹曼加权的构象集合,性能可媲美显式溶剂模拟。本研究使用约200个源自新核磁共振(NMR)实验及文献中的实验观测数据对该方法进行了测试。测试结果表明,该方法在保持极低计算成本的同时,能够以高精度预测溶剂特异性的构象差异,并与实验值的误差控制在一个kBT之内。其高速计算能力(例如可在数分钟内获得一个小分子的构象集合,而非数天)使得该方法非常适用于大规模数据集分析和迭代设计流程。
结果
技术验证
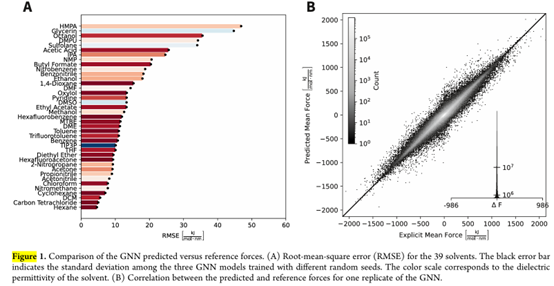
本研究中,图神经网络(GNN)模型在约37万个多样化的小分子(分子量 < 500 Da)上进行了训练,并在一个包含1000个化合物的外部测试集上评估了其准确性,测试集分子量范围为500至700 Da。每种溶剂下的均方根误差(RMSE)如图1A所示。整体而言,GNN的预测结果与外部测试集之间的相关性非常好(图1B)。在不同溶剂中,预测力与参考力之间的偏差变化较大,从己烷中的 4.6 kJ·mol⁻¹·nm⁻¹ 到 HMPA 中的 46.9 kJ·mol⁻¹·nm⁻¹ 不等。除了极性影响之外,其他因素如构象柔性、自扩散性和溶剂的旋转相关时间(即溶剂在某一构象周围达到平衡所需的时间)也可能产生影响,这也可能解释了为何 HMPA、甘油和辛醇的预测误差最大。尽管对于大多数溶剂而言,为获取玻尔兹曼加权的溶剂构象分布所选择的采样时间是足够的,但对于这些较大的溶剂,在测试集计算中可能会出现较大的采样误差。然而,由于GNN是在多个构象上进行训练的,即使在测试集中准确性较低,也未必会在实际应用中(如自由能剖面或构象集合预测)导致显著偏差。因此,在构建训练集时,针对这些较大溶剂的采样时间未作延长。此外,本研究还使用三个不同的随机种子初始化训练了三组模型并进行了评估。总体来看,三者在性能上几乎一致(RMSE的标准差最大不超过 0.05 kJ·mol⁻¹·nm⁻¹)。因此,后续分析统一选用其中一个模型。为了保守起见,选择了在所有溶剂上平均RMSE最高的那个模型。
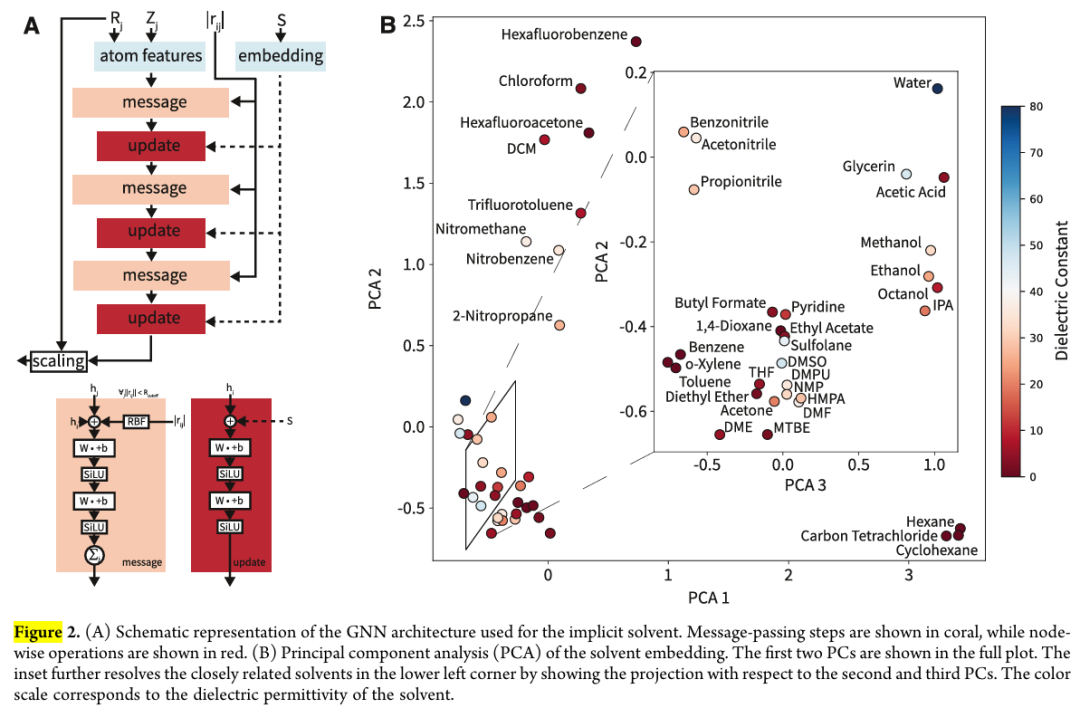
溶剂嵌入(Solvent Embedding)
本研究GNN架构的一个关键点(见图2A)是对所有溶剂集合进行同时训练。简而言之,本研究使用单一模型对整个溶剂集合进行训练,溶剂之间的差异通过溶剂特定的特征向量嵌入(embedding)来体现。这种并行训练策略可以提升信息获取效率,使GNN能够利用迁移学习,即识别并关联表现相似的溶剂与其相关的特征。虽然这种策略意味着每次加入新溶剂时都需重新训练模型,但相比为新溶剂生成训练集的成本,重新训练模型所需的计算代价几乎可以忽略不计。为了探索GNN是否具备发展出一种“人工化学直觉”的能力,本研究提取了GNN中39种溶剂在嵌入层的特征向量,并对其进行了主成分分析(PCA)。图2B展示了前三个主成分的结果,这三者合计解释了47%的方差。所得的三维投影图将具有相似化学性质的溶剂(如具有相同官能团的溶剂)聚集在一起,这不仅符合化学家的直觉,也超越了仅以介电常数为依据的传统溶剂分类方法。例如,四氢呋喃(THF)和二甲基亚砜(DMSO)尽管介电常数差异很大,但由于它们都是极性非质子溶剂,因此在PCA投影中位置相近。相反,一些介电常数相似但官能团不同的溶剂(如氯仿和乙酸)在投影中则相距较远。这一发现表明,GNNIS模型确实能够识别传统连续介质隐式溶剂模型中未能捕捉到的溶剂效应,并验证了本研究同时训练多种溶剂这一策略的有效性。
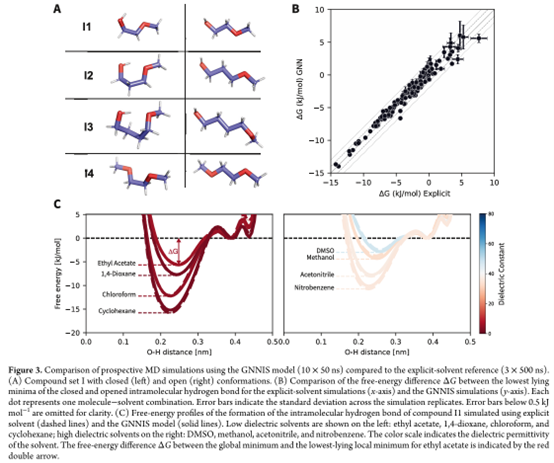
前瞻性分子动力学模拟(Prospective Molecular Dynamics Simulations)
接下来,本研究评估了 GNNIS 模型在预测性分子动力学(MD)模拟中的表现,验证其是否能够重现外显溶剂条件下的 MD 模拟。本研究选用了一个未参与训练集的额外化合物集合 I(见图 3A)进行测试。重要的是,集合 I 中的所有化合物都能够形成分子内以及通过溶剂介导的氢键,因此它们对溶剂效应较为敏感,这些效应并不能简单地通过溶剂的介电常数来解释。
本研究从 GNNIS 模型与外显溶剂 MD 模拟中提取了分子内氢键的自由能分布图以及“开链”与“闭链”构象之间的自由能差值 ΔG。由于自由能差是通过构象分布比例计算获得的,因此模拟中氢键的保持时间、模拟长度和输出频率会影响自由能差的可探测范围。为确保比较的结果不超出该可探测范围,本研究将外显溶剂的参考模拟拆分为三等份进行分析,并对 GNNIS 模拟执行了十次独立重复实验以评估其结果的稳定性。
图 3B 的对比结果表明,GNNIS 与外显溶剂模拟在自由能计算上的一致性极高。图 3C 展示了化合物 I1 在四种低介电常数溶剂(ϵ < 10)和四种高介电常数溶剂(ϵ > 30)中的完整自由能分布。正如预期,即使这些溶剂的介电常数相似,自由能曲线仍存在显著差异,而 GNNIS 模型成功再现了这种差异。相比之下,最先进的隐式溶剂模型 GB-Neck2 与外显溶剂参考结果之间存在显著偏差。因此,且鉴于 GNNIS 模型本身是在 GB-Neck2 框架基础上构建的,后续比较均采用 GB-Neck2 模型作为基准。GNNIS 模型的高精度尤其值得注意,因为其在模拟多个系统时,计算成本远低于外显溶剂模拟。在本研究中,多个体系可以并行进行 GNNIS 模拟,而外显溶剂模拟则需要充分利用现有硬件(如 GPU)来模拟单一体系。GNNIS 模拟不依赖于单个模拟充分利用 GPU,因此能在同一硬件平台上并行处理多个体系,实现近 10 倍加速(在 8 核 CPU + 1 张 NVIDIA RTX 4090 GPU 上,模拟速度为 1750 ns/天 对比 16900 ns/天)。
快速评估构象分布
本研究的一大优势在于:基于隐式溶剂的模拟方法可以直接估算构象的溶剂化自由能,而这在显式溶剂模拟中则需要代价高昂的自由能计算。因此,隐式溶剂模型可用于识别构象能量面上的极小值,并计算其相应自由能,从而快速获取玻尔兹曼加权的构象分布。传统隐式溶剂模型生成的构象分布准确性有限,主要因为它们难以描述显式溶剂效应。相比之下,如上文所示,本研究所提出的 GNNIS 模型能够准确再现这些显式溶剂效应。本研究进一步探索了该方法在快速识别小分子玻尔兹曼加权构象分布方面的可行性。相关流程如图 4A 所示(详见方法部分)。简而言之,首先通过距离几何方法随机生成多样化构象集合,采用标准力场(OpenFF 2.0.0)并结合 GNNIS 模型进行最小化。然后根据每个构象的势能进行排序,并根据均方根距离(RMSD)进行剪枝。最后一步使用 GNNIS 预测的势能,并结合 Grimme 的 quasi-RRHO 熵估算法计算最终构象的自由能,其中所有模式的熵会被阻尼,并补充自由转子的熵贡献。
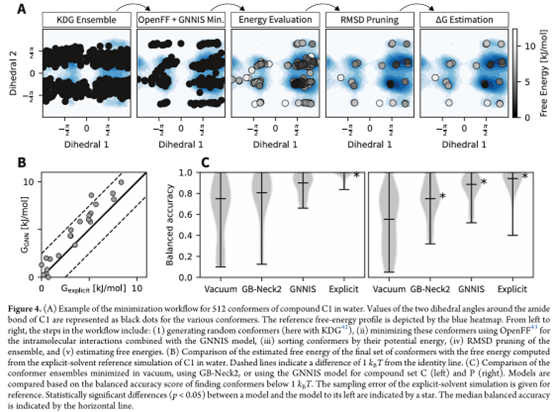
模拟构象分布
本研究在两个小分子数据集 C 和 P 上进行评估,其中 C 集合包含分子量较小的分子(< 500 Da),并将 GNNIS 预测的构象分布与显式溶剂模拟结果进行了比较。如图 4B 所示,针对一个在水中模拟的分子,GNNIS 预测的构象分布与显式溶剂参考模拟结果整体一致。为了定量评估构象分布的准确性,本研究计算了使用 OpenFF 2.0.0 最小化的不同模型的“平衡准确率”,包括:(i)无溶剂(真空)、(ii)GB-Neck2 隐式溶剂模型,以及(iii)GNNIS 模型,均与显式溶剂模拟的结果进行比较。低能构象的正确预测尤其重要,因为它们对实验可观测数据(如 NMR 或 IR 谱)具有显著贡献。如图 4C 所示,GNNIS 显著优于 GB-Neck2,在识别低能自由能极小值方面具有更高准确性。在数据集 P 中,GNNIS 模型的最小化结果甚至接近显式溶剂模拟(50 ns 和 100 ns REST2 模拟)的平衡准确率。由于数据集 P 的分子相比 C 更大且更具柔性,在显式溶剂中需更长的模拟时间以提升准确性。
这些结果非常具有前景,因为该流程的计算开销远低于显式溶剂模拟。
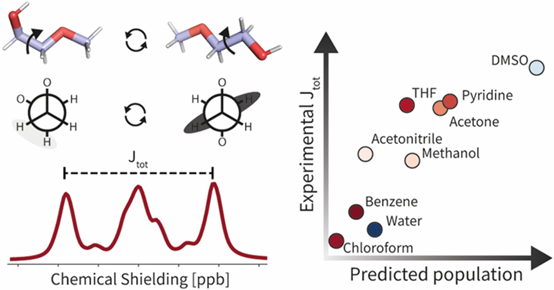
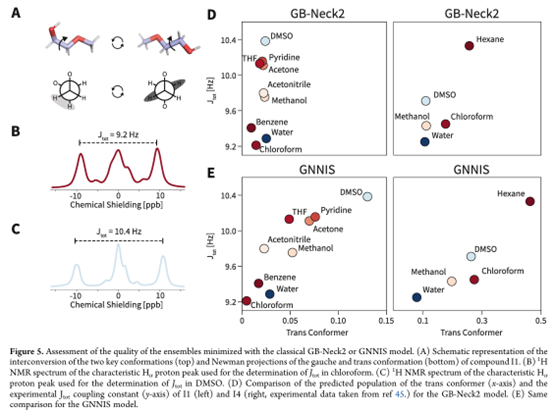
与实验可观测数据的比较
为了进一步验证本研究构象分布在实际应用中的有效性,本研究按照上述流程对化合物 I1 和 I4(图 3A)进行了处理,这两者均在具有高分辨率质子 NMR 谱数据的溶剂中进行了分析。在室温下,I1 和 I4 都具有两个主要的构象,可以相互转化(见图 5A)。其相对构象的分布可通过甲氧基上的 α-质子的 J 偶合常数推断得出。Hα 的 NMR 信号为多重峰,由两个不同的近邻质子–质子 J 偶合(JA−B 和 JA−C)引起。JA−B 和 JA−C 是所有构象的旋转平均值。本研究中提取了总标量偶合常数 Jtot = JA−B + JA−C,即多重峰最外侧两个峰之间的频率差。在氯仿中,gauche 构象占主导,从而导致 Jtot 最小(见图 5B);而在 DMSO 中,由于 trans 构象比例增大,Jtot 明显升高(见图 5C)。图 5D 和 5E 展示了 GNNIS 和 GB-Neck2 分别预测的 trans 构象比例与实验 Jtot 之间的相关性。对于两个化合物,采用 OpenFF 2.0.0 和 GNNIS 最小化的构象集合均显示出 trans 构象占比与 Jtot 呈线性关系,显示出本研究所提出流程能够快速生成合理的溶液态构象分布,显著优于 GB-Neck2 模型。一个有趣的发现是,化合物 I1 的开链与闭链构象的相对比例与“极性溶剂更易促进开链构象”的传统观点不符(GB-Neck2 模型确有此趋势)。这可归因于 GB-Neck2 仅依赖固定介电常数的连续介质模型来预测构象分布。例如水、DMSO 和甲醇由于介电常数较高,GB-Neck2 预测它们具有最多的 trans 构象,而苯(最低介电常数)则最少。但实际上,对于 I1 和 I4,在水中反而观察到 trans 构象的比例最低。不过,对于化合物 I1 的丙酮腈与 THF 两个溶剂(图 5E 左图),GNNIS 模型预测结果偏离了实验趋势。但显式溶剂模拟也显示出类似的 trans 构象占比(丙酮腈 2.9%,THF 6.0%),与 GNNIS 最小化结果(丙酮腈 2.3%,THF 4.9%)基本一致,说明该偏离可能来源于显式溶剂模型本身,而非 GNNIS。本研究同时指出,即使存在溶质力场偏差,也应影响所有溶剂而非仅在某一特定溶剂上出现异常。
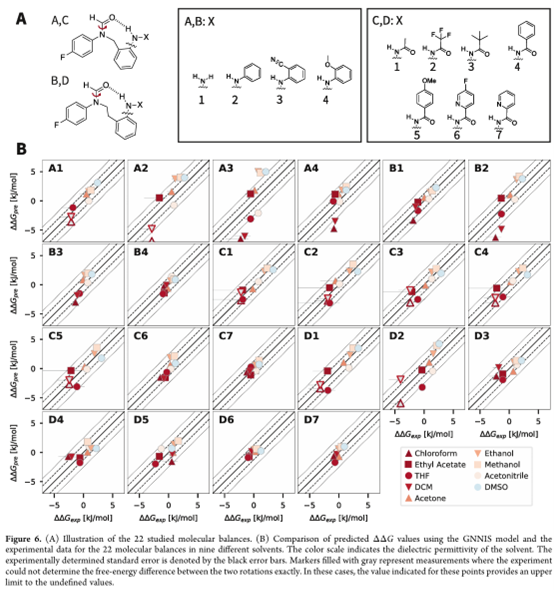
进一步的验证:分子平衡实验
为进一步验证所提最小化流程的适用性,本研究研究了 22 个“分子平衡体”(图 6A),这些分子可用于量化不同溶剂中的氢键强度。Meredith 等人 对这些化合物在九种溶剂中通过 ¹⁹F{¹H} NMR 测量了两个酮基旋转构象的分布,从而反映了分子内氢键强度及构象分布。本研究对这些分子在各个溶剂中采用本研究的最小化流程进行分析。研究重点在于:是否能准确捕捉不同溶剂环境中,构象之间微小的自由能差异。具体地,本研究首先基于最小化后的构象集合预测两个酮基旋转态的自由能差(ΔG),然后计算其相对于均值的偏差(ΔΔG_pre),并与实验测得的自由能差偏差(ΔΔG_exp)进行比较。结果如图 6B 所示,GNNIS 能够准确地再现不同溶剂中构象分布的差异,大多数 ΔΔG 值都在 1kBT 以内。尤其值得注意的是,该旋转过程根据实验发生在微秒时间尺度,在显式溶剂中进行分析代价极高。而 GB-Neck2 模拟结果显示,不同极性溶剂下几乎预测相同的构象分布,明显低估了溶剂效应的强度。这些结果再次表明,本研究提出的 GNNIS 模型在准确性和计算效率之间实现了良好平衡,特别适合研究此类体系。
结论
本研究将 GNNIS 方法扩展至 39 种有机溶剂,并提出了一种结合标准力场、能够快速识别小分子在溶液中 Boltzmann 加权构象分布的工作流程。GNNIS 模型在涵盖 39 种不同溶剂的多样化小分子模拟数据集上进行了训练,随后通过前瞻性分子动力学模拟进行了验证,并在基于 NMR 实验确定的构象偏好上进行了测试。两类对比均显示出本研究方法与参考数据之间具有极高的一致性。
此外,与显式溶剂模拟相比,本研究方法大幅提升了计算效率(例如,构象分布的生成时间从小时级缩短至分钟级),使其成为理解溶液中分子行为的有力工具。这一进展有望促进分子的理性设计,支持更大规模数据集的研究和更快速的实验反馈,同时实现对实验可观测量的精准且高效的解释。
整理 | 刘名权
参考资料
Katzberger, P., Hauswirth, L.M., Kuhn, A.S., Landrum, G.A. and Riniker, S., 2025. Rapid Access to Small Molecule Conformational Ensembles in Organic Solvents Enabled by Graph Neural Network-Based Implicit Solvent Model. Journal of the American Chemical Society.
内容中包含的图片若涉及版权问题,请及时与我们联系删除

评论
沙发等你来抢