
DRUGAI
机器学习原子间势(MLIPs)已成为现代原子模拟的重要工具。近期,基于大规模数据集预训练的通用MLIPs展现出优异的精度与泛化能力。然而,其计算成本仍限制了在化学无序系统(需大尺寸模拟单元)或采样密集型统计方法中的应用。研究人员在本研究中引入了连续且可微的炼金自由度,利用图神经网络MLIPs中将元素表示为实值张量的特性。该方法在输入图中加入具有权重的炼金原子,并调整了MLIP的消息传递与读出机制,从而实现材料组成状态之间的平滑插值。借助MLIP的端到端可微性,研究人员可高效计算能量对组成权重的梯度。据此,提出了用于优化固溶体组成以实现目标宏观性质、解析多组分氧化物的有序与无序结构,以及开展炼金自由能模拟以量化空位形成与成分变化自由能的方法。

研究结果
炼金图构建与消息传递机制
研究人员在不更改已有模型参数的前提下,对图神经网络结构进行修改,以支持材料中不同组分的连续建模。在标准的机器学习原子势(MLIP)中,原子系统被表示为图结构,其中原子为节点,相邻原子对形成边。每种元素通过嵌入向量初始化节点特征,边特征由相对位移决定,并通过多层消息传递机制传播信息,最终预测体系总能量。
为引入组分变化,研究人员提出将“炼金原子”加入图结构中。以LiCl、NaCl、KCl混合为例,通过分配相应的炼金权重(如20%、30%、50%),原始节点可被拆分为多个对应元素的节点。这些节点继承相同的原子位置,并通过修改消息传递与能量读出机制,实现对不同组分状态间的平滑插值。整个过程保持端到端可微,并确保在极端组分(如全为某元素)下预测能量与原始图一致。
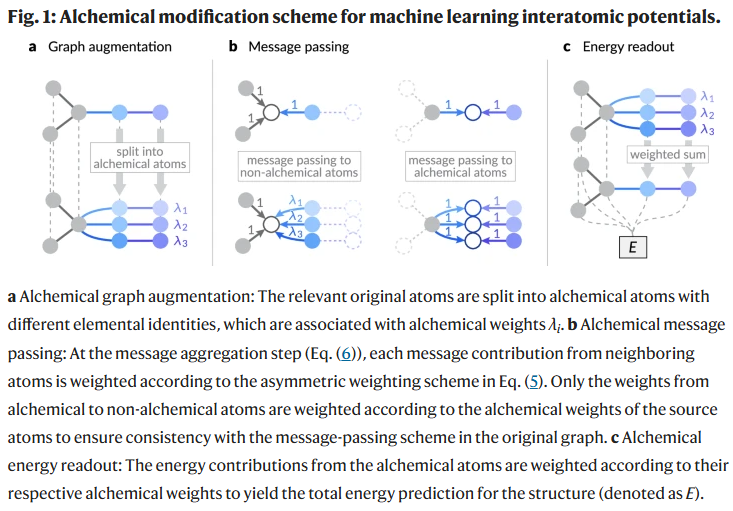
固溶体建模能力
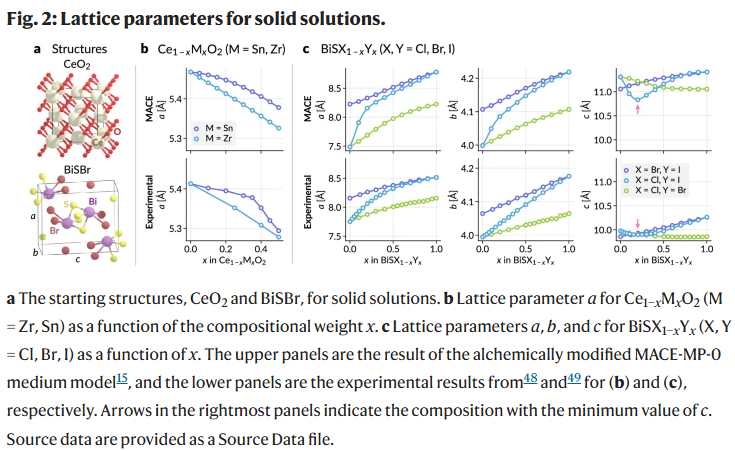
为验证炼金图在固溶体建模中的能力,研究人员以晶格参数为测试指标,评估其是否符合Vegard定律或存在偏离。在Ce₁₋ₓMₓO₂(M=Zr, Sn)体系中,炼金模型成功再现了Zr体系中的线性变化以及Sn体系中出现的正偏离。类似地,BiSCl₁₋ₓIₓ体系中的晶格常数a和c表现出与实验一致的非线性趋势,特别是c参数的极小值位置预测准确,尽管绝对值存在偏差。
可微分组分优化
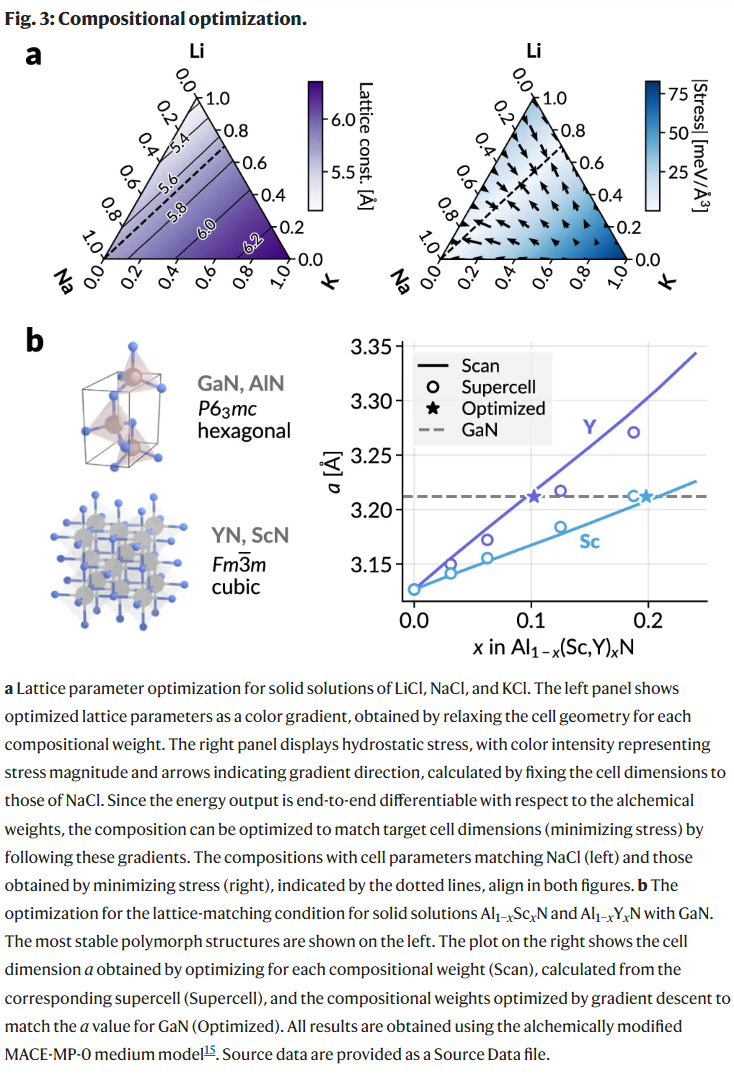
利用炼金权重的可微性,研究人员进一步开展了组分优化实验。首先在LiCl-NaCl-KCl混合体系中,通过对应力的导数进行梯度下降,可有效寻找使晶格常数匹配目标值的最优组分组合。在更复杂的体系如Al₁₋ₓScₓN和Al₁₋ₓYₓN中,通过调节炼金组分及晶胞参数,实现与GaN匹配的应力最小化组分,所得结果与扫描法和实际掺杂模型高度一致,表明炼金方法可作为具有代表性的紧凑建模手段。
无序体系热力学建模
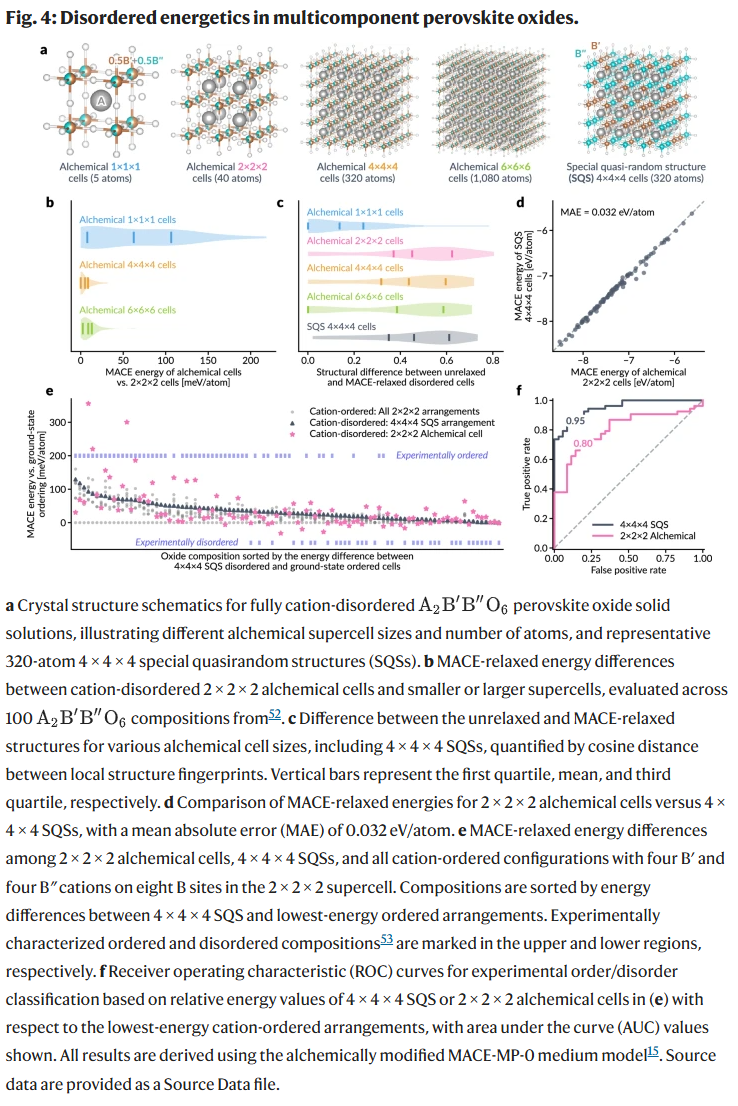
为了进一步验证方法在无序固溶体建模中的可扩展性,研究人员选取多组分钙钛矿为测试对象,对比炼金超胞与传统特征无序结构(SQS)在建模精度和计算效率上的差异。结果表明,使用2×2×2的炼金超胞即可逼近SQS的能量结果,能量均方误差仅为0.032 eV/atom,且能有效区分实验上有序与无序结构。
尽管较小的炼金单胞无法充分表达局部结构畸变,如八面体扭曲等,但中等尺寸(如40原子的炼金超胞)已足以用于分类与趋势预测。相比之下,炼金模型无须SQS方法中的模拟退火与大超胞构建,显著提升了高通量筛选的效率。
本方法在几何假设上类似于虚拟晶体近似(VCA),即采用平均化结构描述固溶体,但不同于VCA对赝势强依赖的“赝势炼金术”,炼金MLIP方法以图神经网络为基础,通过训练内建正则化保持结果物理合理,避免了繁琐的手动调参。
自由能计算
研究人员采用非平衡切换方法计算自由能,其中哈密顿量依赖于一个从 0 到 1 变化的参数 λ,用于连接初始与最终态。在等体积等温系综下,系统在有限时间内从 λ = 0 切换到 λ = 1,会产生不可逆功,进而得到自由能差。通过自动微分,研究人员直接利用炼金权重计算能量对 λ 的梯度,相比传统的线性插值更高效,因其避免了对不变原子的重复计算。具体计算效率与自由能结果的比较详见补充材料。
空位形成自由能
研究人员以体心立方铁中的空位为例,计算其低温下的吉布斯自由能,并与常用的 Frenkel–Ladd 方法进行比较。通过引入炼金路径,将空位原子的炼金权重从 1 逐渐变为 0,实现该原子的“炼金消失”,并同步引入谐振子势能,将完美晶体平滑转化为含空位晶体与一个谐振子。结果表明,炼金方法得到的空位自由能与参考方法相当,且标准差更小,尤其在很短的切换时间内仍具有良好收敛性和统计稳定性。
炼金自由能与组分变化
为了评估炼金方案在组分变化下自由能计算的能力,研究人员以卤化物钙钛矿 CsPbI₃ 和 CsSnI₃ 为例,比较其钙钛矿相与非钙钛矿相之间的自由能差异。通过构建炼金路径,将Pb原子逐步转化为Sn原子,研究人员计算了组成变化所引起的自由能差,并与 Frenkel–Ladd 方法进行对比。结果显示,两种方法在高温下表现一致,而在低温下存在轻微偏差,可能源于不同模拟条件下发生的晶相转变。
此外,在400K条件下,研究人员分析了炼金路径在非平衡切换模拟中的自由能收敛性,结果再次证明该方法比传统路径收敛更快,能量耗散更小。由于两种不同组分但结构相似的材料在相空间中的重叠较大,炼金路径能显著提升自由能模拟的效率。
讨论
本研究提出的炼金MLIP修改方法,使得具有不同组成的结构之间能够实现平滑插值。在原始MLIP原型的基础上,研究人员对输入图结构、消息传递机制和读出层进行了调整,引入炼金权重来表达多组分状态。该方法可泛化至多种MLIP架构,尤其在与MACE模型结合时效率更高,因其特有的基于双体消息构建多体特征的方式。
研究人员首先将该方法应用于固溶体建模。尽管炼金权重与化学计量之间不存在严格的理论对应关系,结果表明该方法能够捕捉部分固溶体晶格参数的非线性偏离。此外,该方法的可微性支持组分优化,以实现目标晶格常数匹配。在多组分钙钛矿中,该方法还能以更小原子数、无需结构优化的方式实现对有序与无序行为的准确建模,其效果可与SQS相媲美。
更进一步,炼金权重的引入使得原子类型的平滑变换、甚至原子的创建与湮灭成为可能,从而支持不同组分状态之间的自由能差计算。研究人员展示了该方法可高效评估体心立方铁中空位的自由能,以及CsPbI₃和CsSnI₃钙钛矿与非钙钛矿相的相对稳定性,相较于传统Frenkel–Ladd路径,该方法具有更高效率。需注意,尽管固溶体建模依赖近似假设,炼金自由能计算在理论上是精确的,只要达到收敛条件。
总体而言,该方法可在不改动底层MLIP结构的前提下,实现在组成变化下的高效建模。其主要误差来源并非来自炼金框架本身,而是来自MLIP模型的准确性,包括:(1) MLIP与DFT之间的误差;(2) DFT本身的近似误差。鉴于大多数通用MLIP是在结构弛豫轨迹附近的数据上训练的,因此在能量极小值附近误差较小。若将模型在相关组成空间上的DFT数据进行微调,可进一步减小上述误差。此外,也可引入自由能扰动方法,从更精确的哈密顿量中计算自由能,以纠正这两类误差。
研究人员还指出,可通过可微模拟框架对MLIP进行进一步优化,使其输出的晶格参数或自由能更贴近目标值,以提高模型的物理一致性。 尽管本工作中炼金权重用于表达多组分原子占据情况,未来工作也可探索将λ与原子序数直接耦合的方案,从而与量子炼金研究接轨。尽管当前的预训练嵌入未必理想,但通过炼金力匹配或基于分析梯度的微调,有望建立与量子炼金一致的MLIP表达,形成面向热力学积分计算的物理合理近似。展望未来,炼金自由度在MLIP中不仅可用于本研究展示的应用场景,其对材料和分子的生成建模也具有潜在价值。结合离散采样策略,研究人员预计此类自由度将在多组分材料与分子设计任务中发挥更广泛作用。
整理 | WJM
参考资料
Nam, J., Peng, J. & Gómez-Bombarelli, R. Interpolation and differentiation of alchemical degrees of freedom in machine learning interatomic potentials. Nat Commun 16, 4350 (2025).
https://doi.org/10.1038/s41467-025-59543-2
内容中包含的图片若涉及版权问题,请及时与我们联系删除

评论
沙发等你来抢