
DRUGAI
蛋白质中的内在无序区(IDRs)在细胞功能中发挥着关键作用。越来越多的研究表明,IDRs 往往以不依赖氨基酸精确排列的方式与作用对象结合,而是通过化学互补性驱动相互作用,形成无序的结合复合物。然而,这类具备化学特异性的动态相互作用极难预测。
在本研究中,研究人员借用最初为分子模拟开发的化学物理原理,构建了一种全新方法,利用蛋白质序列作为唯一输入来预测 IDRs 与其相互作用蛋白之间的化学特异性。该方法被命名为 FINCHES,能够直接预测相图、识别 IDRs 上的化学特异性互作热点、分解 IDRs 中不同的化学功能结构域,并为探索 IDRs 在分子识别过程中的机制提供假说与验证路径。

蛋白质中的内在无序区(IDRs)在各类生物中广泛存在。与具有稳定三维结构的折叠结构域不同,IDRs 通常以多种构象共存的异质状态存在。尽管缺乏固定的三级结构,这些无序区域在多种不同的细胞过程中发挥着关键作用,其功能往往依赖于与多种作用对象之间的分子互作。
部分 IDRs 在结合过程中会折叠形成有序结构,但在许多情况下,即使处于结合状态,其结构仍保持一定程度的无序性,从而形成所谓的“模糊互作”。IDRs 可以通过两种主要方式与作用对象结合:序列特异性识别和化学特异性识别。
在序列特异性识别中(也称为位点特异性识别),IDR 在结合后形成一定程度的有序结构,与作用对象构建出相对传统的结合界面,尽管这种界面可能是短暂的。而在化学特异性识别中,结合状态缺乏稳定结构,吸引作用主要依赖于相互间的化学性质互补,使得结合构象呈现出多样化的集合状态。
虽然近年来基于结构信息训练的深度学习模型在识别序列特异性互作方面表现出色,但在解析化学特异性的分子识别方面仍存在显著局限。
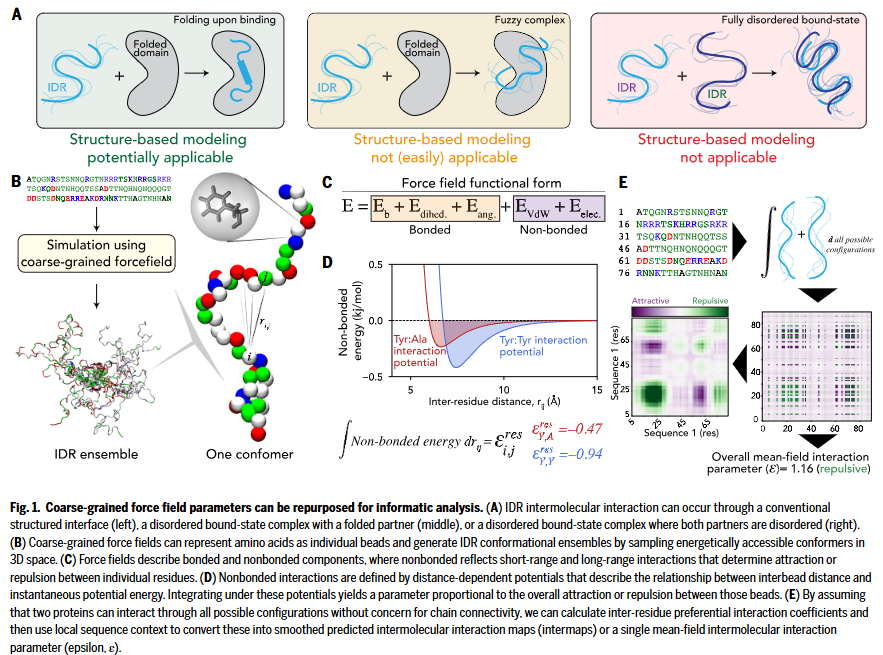
分子力场在 IDR 分析中的重构与利用
分子力场通过一系列公式与参数来描述生物分子的化学物理特性。近年来,针对无序蛋白的粗粒化建模推动了多个力场的发展,如 Mpipi 和 CALVADOS 系列,已被证实可准确预测 IDRs 的整体尺寸特征。这些模型通过定义氨基酸对之间的非键合相互作用势,定量描述任意距离下残基之间的吸引或排斥作用。
研究人员认为,这一套原本用于分子模拟的化学物理体系可以被提取并改造:通过对残基对势能函数的积分,计算出平均场互作参数,类似于不含体积修正项的 Mayer f 函数。在此基础上,结合带电残基与脂肪族疏水残基的局部环境特征,对个别氨基酸的互作强度进行调节,最终生成残基对之间的互作矩阵。该矩阵可以整体平均,得到两个序列之间的平均场互作参数 ε,或通过滑动窗口局部平均,用于识别吸引或排斥的互作区域。
研究人员将这种预测的分子互作图谱命名为 “intermaps”。其中 ε 的值为负表示吸引互作,为正表示排斥互作。
该方法的一个核心假设是:IDRs 之间的吸引作用完全来源于化学互补性,即在遍历所有可能构象的极限下所显现的化学特异性,而非特定子区域之间精准的结构化互作。此外,该方法不考虑不同区域间的协同作用,这也带来一定的局限性。
尽管如此,该方法仍可高效计算两条序列之间的平均场互作参数,并识别出可能驱动吸引或排斥作用的局部片段。与深度学习方法不同,该方法的分子互作机制完全可解释,其决定因子由力场的函数形式与参数明确定义。
本研究在 FINCHES 工具中使用了 Mpipi-GG 和 CALVADOS2 两种力场,同时也支持接入其他力场模型。这些力场通过引入 Debye–Hückel 项,可调节溶液中盐浓度以研究盐依赖性互作效应。此外,模型具备高通量性能,在一个 100 个残基长度的 IDR 上,每秒可预测超过 1000 条序列。
研究人员强调,虽然所得互作得分无法提供高分辨率的定量预测,但足以进行快速的半定量分析,用于评估 IDRs 潜在的分子互作趋势。
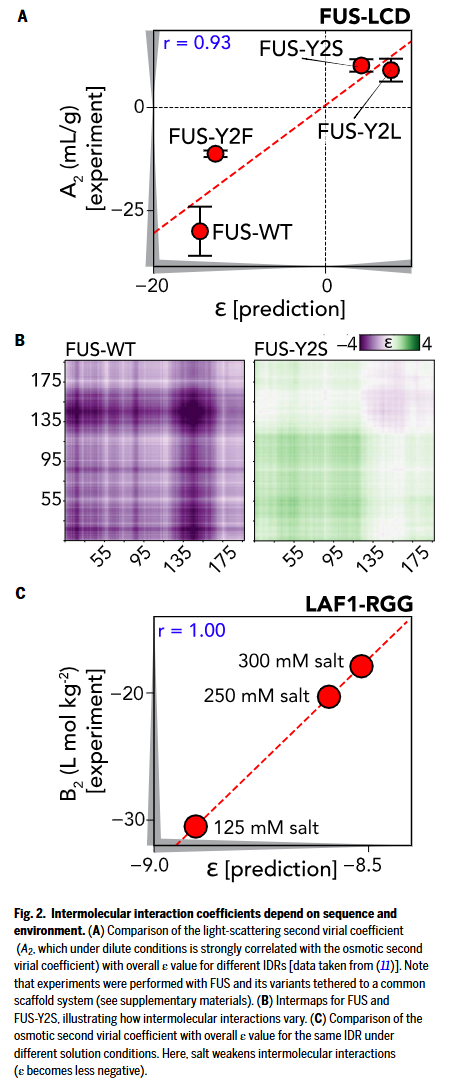
与实验相互作用数据的验证
研究人员首先探究了计算得到的平均场互作参数(ε)是否与实验可测的分子间互作强度呈比例关系。渗透压和光散射实验中测得的二级维里系数(分别为 B₂ 和 A₂)反映了体系偏离“理想非互作”状态的程度:负值表示总体吸引作用,正值表示总体排斥作用。研究人员选取了两个已有二级维里系数测定数据的 IDR 体系进行分析:分别是 RNA 结合蛋白 FUS 的低复杂性结构域变体,以及 DEAD-box 解旋酶 LAF-1 的 RGG 结构域。对于 FUS,研究人员计算了一系列突变体的 A₂ 值,结果与预测的 ε 值高度相关。尽管两者量纲不同,但二者的零点应具有等效性,这一预测得到了最佳拟合线通过原点(0,0)的验证。
此外,将野生型 FUS 与酪氨酸全部突变为丝氨酸的突变体(Y2S)进行 intermap 比较,显示其吸引互作被完全抑制。对于 RGG 结构域,研究人员计算了不同盐浓度下的 ε 值,并与 NaCl 浓度依赖的 B₂ 实验值进行比对,结果呈现 1:1 的高度对应关系。尽管在将二级维里系数与 ε 值对应时存在一定假设与局限,但整体趋势增强了研究人员对其理论假设合理性的信心。
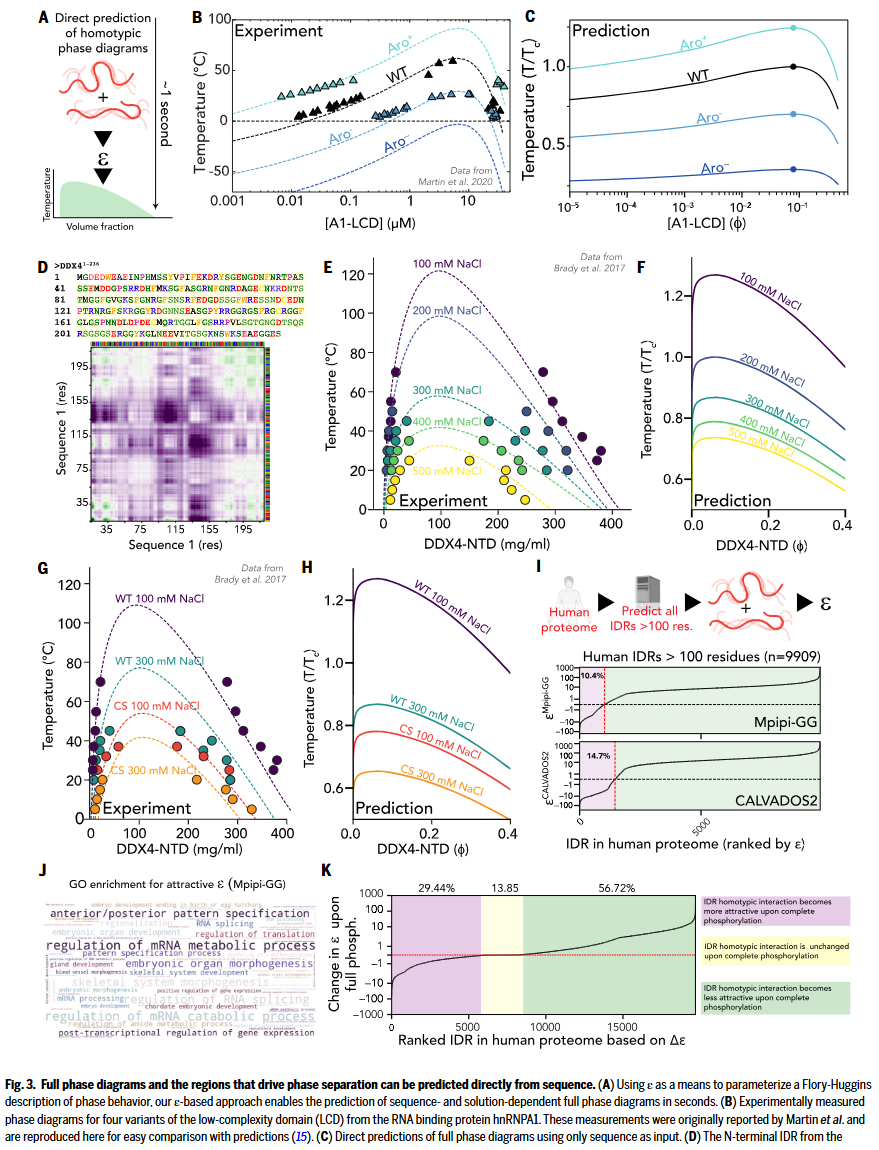
从序列直接预测相图
研究人员开发的方法可直接从蛋白质序列预测相图,为解析 IDR 介导的相分离行为提供了新路径。通过将预测得到的同源互作参数 ε 值引入 Flory-Huggins 理论,并转换为 Flory χ 参数,研究人员利用解析解快速生成多种无序蛋白体系的相图,能够定性再现序列和环境变化如何影响相行为。随机打乱序列的对比分析表明预测结果依赖于氨基酸排列顺序。以 hnRNPA1 芳香族残基突变体为例,该方法准确再现不同突变体相图的变化趋势,显著缩短了以往依赖模拟与实验的预测流程。进一步测试显示,该方法可捕捉序列图案、带电残基种类及溶液条件对相行为的影响。以 DDX4 为代表的序列在不同盐浓度或突变处理下的相图预测,与已有实验数据高度一致。研究人员还在 hnRNPA1、FUS、RLP、LAF-1 和 TDP-43 等多个体系中验证了该方法的广泛适用性,准确复现了芳香/疏水残基组成、带电残基位置、淀粉样区域及残基密度对相分离的调控作用。在人类蛋白质组层面,研究人员评估了所有长 IDR(>100 个残基)的同源 ε 值,发现 10–15% 的 IDR 显示出潜在吸引性互作,且这些蛋白显著富集于 RNA 相关、形态发生及发育功能中。intermap 可进一步揭示具体序列驱动因素,例如 HAX1 蛋白。此外,研究人员还系统评估了翻译后修饰的调控作用,模拟磷酸化修饰(将 Ser/Thr/Tyr 位点转为 Glu)后,约 57% 的 IDR 显示吸引性下降,约 30% 上升。例如 ZO1 蛋白的 C 端 IDR,在磷酸化后预测出显著的互作减弱,符合已有实验证据。这些结果表明,IDR 的化学特异性互作不仅由序列本身决定,也可通过翻译后修饰灵活调控。
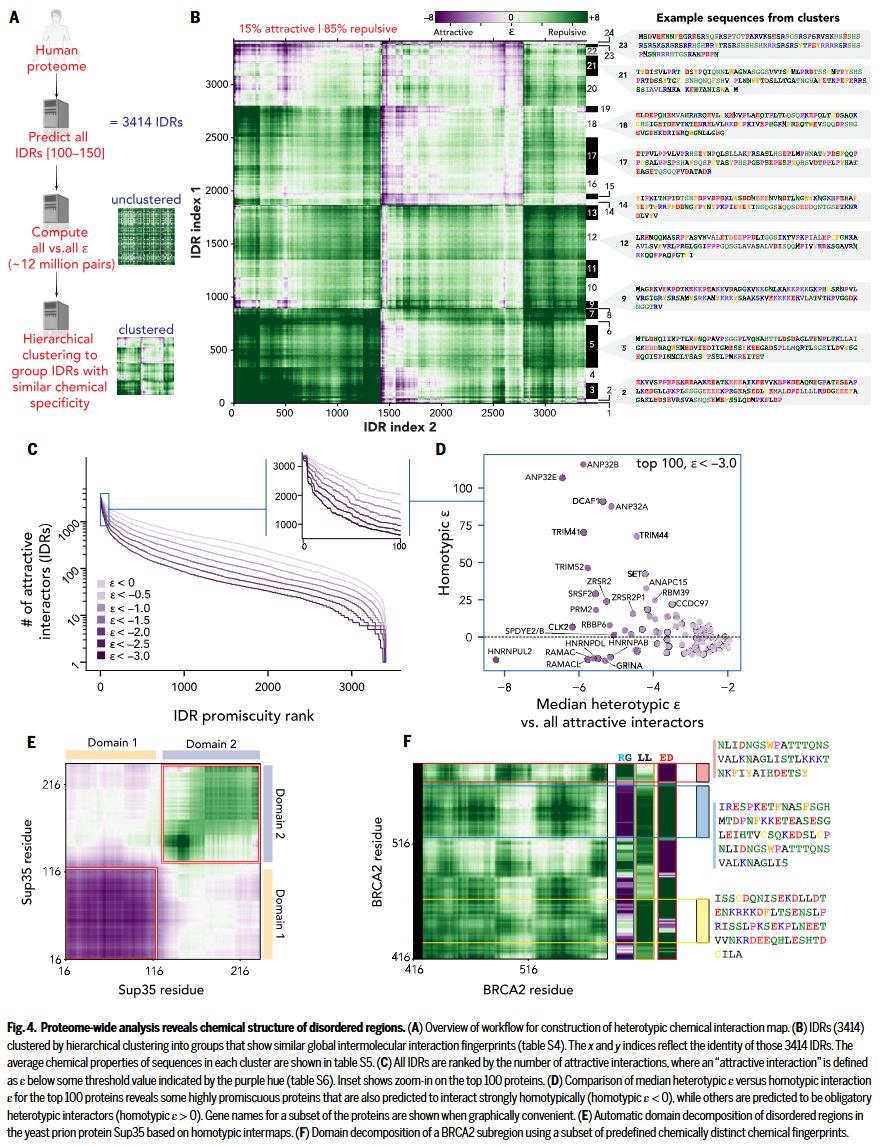
分子互作化学空间中组织 IDRs
鉴于内在无序区(IDRs)在序列比对中通常表现出较低的保守性,研究人员进一步探讨了是否可以基于化学互作特征(而非序列相似性)对 IDRs 进行分类。他们从人类蛋白质组中筛选出长度在 100 至 150 个残基之间的所有 IDRs(共 3414 条),以确保互作配对在长度上相对均衡,并计算了所有可能的 IDR 两两互作(约 1200 万组),从而构建了蛋白质组尺度的异源互作图谱。通过层次聚类,研究人员将这些 IDRs 分为 24 个化学互作指纹相似的簇,尽管它们在一级序列上高度多样。值得注意的是,仅有约 15% 的 IDR-IDR 异源互作显示出吸引性,大多数为排斥性。虽然整体 ε 值为正不意味着 IDRs 一定无法互作,但这些结果凸显两个关键点:其一,在平均场分子互作层面,自然界中的 IDRs 可划分为不同的化学特性“生态位”;其二,序列所决定的化学性质影响一个 IDR 是更偏好同源还是异源吸引互作,而某些化学组合则表现出更强的“化学多配性”。为进一步研究这种“多配性”,研究人员将其定义为一个 IDR 对多种不同作用对象均呈现吸引 ε 值的倾向。通过统计每个 IDR 的异源吸引互作数量,发现许多 IDRs 具备高度多配潜力。在多配性最高的前 100 个 IDRs 中,相关蛋白涵盖 RNA 结合、细胞稳态维持、细胞凋亡、转录调控与组蛋白伴侣功能等多个分子过程。此外,结合蛋白丰度信息分析发现,高表达且具多配性 IDR 的蛋白几乎全部为 RNA 结合蛋白。尽管大多数 IDR–IDR 配对为排斥性,但每个 IDR 至少与某一个 IDR 具有吸引性互作,而部分 IDRs(如聚类簇 23 和 24)则表现出广泛的吸引性互作。更进一步,这种基于分子互作化学特性的 IDR 分类策略不仅适用于全蛋白组水平,也可用于个别蛋白内部的功能子结构划分。尽管当前识别 IDRs 本身已较为成熟,但如何进一步对其内部进行功能性亚区段划分仍具挑战,尤其因为 IDR 的功能具有上下文依赖性。若将化学特异性互作视为功能依据,则可以相对于特定互作对象对 IDR 内部进行亚结构域划分,为后续的结构删除实验提供比传统人为裁剪更清晰的指导。例如,在酵母朊蛋白 Sup35 的 N 端或高无序结构蛋白 BRCA2 中,研究人员均展示了该方法可用于识别构成偏倚明显或互作对象未知的 IDR 子结构域。
解码 IDR 介导互作的化学特异性
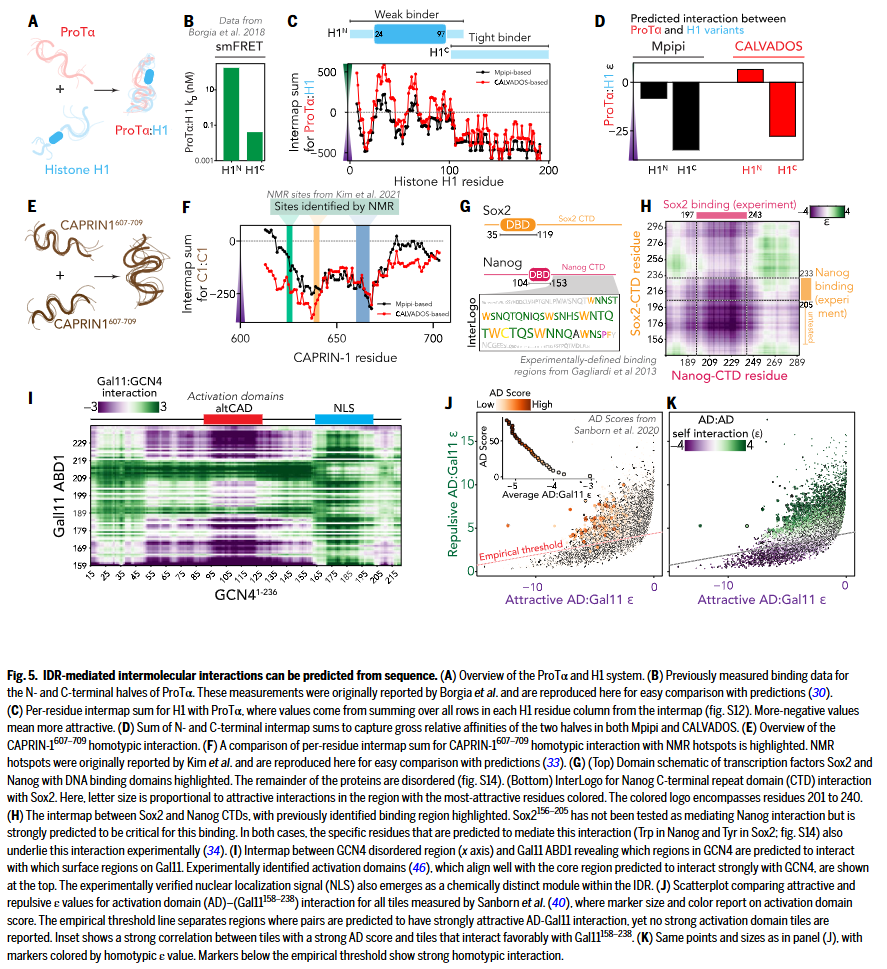
研究人员利用 FINCHES 框架,系统评估了 IDR 与其作用对象之间的化学特异性互作,验证其在解析具体生物物理机制中的适用性。例如,Prothymosin α(ProTα)与组蛋白 H1 可形成完全无序的高亲和力复合物,smFRET 实验数据显示,H1 的 C 端无序区与 ProTα 的结合力远高于其 N 端。FINCHES 预测出的 intermap 强度与这一实验趋势高度一致,且可揭示盐浓度对互作的调节效应。另一个案例是应激颗粒相关蛋白 CAPRIN-1,其 C 端 IDR 的同源互作在高盐环境下增强,与 NMR 实验结果一致,所识别的三个位点中有两个在序列层面明确可预测。此外,研究人员还对转录因子 Sox2 与 Nanog 的 C 端 IDR 互作进行分析,成功识别出关键互作残基区域,并使用“InterLogo”将互作强度可视化映射于氨基酸序列上。这些结果表明,即使是低复杂性、高亲和力的互作,其局部化学特异性也可从序列中有效预测。
研究人员进一步探讨了 IDR 与折叠结构域之间的互作预测。以酵母转录因子 GCN4 与共激活因子 Gal11 的结合为例,FINCHES 能准确识别 Gal11 IDR 中与激活功能相关的区域,结果与实验验证的激活域高度重合。在更大规模的数据集中,研究人员对 Sanborn 等人筛选的 7577 条 40 残基片段进行了分析,发现 IDR–Gal11 平均互作强度与其激活功能评分(AD score)呈正相关。尽管部分 IDRs 与 Gal11 显示出强吸引力,却表现出较弱的激活功能,FINCHES 预测这些 IDRs 也具有强烈的同源互作趋势,可能因发生自组装或“离靶”互作而抑制了与 Gal11 的结合,影响转录活性。这一现象与酸性暴露模型的解释一致,并为解读凝聚体形成与转录输出之间的关联提供了理论支持。总体而言,这些结果说明 FINCHES 能够仅依赖序列信息,准确解析 IDR 的分子功能机制,为研究无序区介导的分子识别提供了强有力的工具。
结论
通过提取原用于分子模拟的化学物理原理,研究人员将其解析形式和参数重构用于蛋白质序列层面的计算,从而实现在无结构信息的条件下,直接预测 IDR 的吸引性与排斥性分子互作。虽然该方法存在一定局限性,不应夸大其精度与普适性,但它简洁、高效,支持在蛋白质组尺度上进行互作预测。
FINCHES 专注于预测化学特异性互作,而非结构驱动的序列特异性互作。后者依赖于明确的结构化结合界面,更适用于 AlphaFold、RoseTTAFold 等结构中心方法。此外,该方法不考虑远端残基之间的互信息,也未建模分子内与分子间互作之间的竞争,尽管可通过同时分析同源与异源互作来部分补偿。
FINCHES 的预测建立在粗粒度分子力场(如 CALVADOS2 和 Mpipi-GG)之上,这些模型虽被广泛验证,但仍存在不足,例如对脂肪族疏水作用的低估、无法捕捉由二级结构引发的效应,以及对盐效应和温度依赖行为(如疏水作用)处理不充分。不过这些问题理论上可通过重新参数化模型加以改进,FINCHES 也可作为诊断和优化力场的工具,其开放式架构支持新参数与新模型的即时接入。
研究人员强调,异源互作 ε 值的预测结果不应直接视为体内互作图谱,因为真正的分子互作依赖于结合位点的数量与可结合状态下的蛋白浓度。FINCHES 更适合分析已知互作蛋白之间的序列互作机制,可用于识别关键残基并辅助突变实验验证。
此外,该方法可用于从序列直接预测 IDR 的同源相图,在无需推导通用原则的前提下,实现针对特定突变的定性预测。相比于机器学习方法需依赖数量有限、条件异质的实验数据,FINCHES 更适合在控制变量明确的前提下分析特定互作事件。
在与现有研究相互补充的同时,FINCHES 开辟了多种前景广阔的应用方向。首先,它可用于指导和解释 IDR 介导的分子互作实验,作为连接序列与互作机制的高效假设生成器。其次,该方法可用于解析 IDR 的保守性与功能注释,通过预测其生物物理属性和化学特异性,在一级结构保守性低的情况下提供功能线索。第三,该方法支持基于互作轮廓的上下文依赖型结构域划分,尤其适用于一个 IDR 与多个配体存在不同互作残基的场景。第四,FINCHES 的通量与可拓展性为理性设计 IDR 提供了便利,例如调控相分离行为、优化结合位点亲和力与特异性,或通过 IDR 设计干预分子间排斥,实现熵驱动功能控制。相关设计工具包 GOOSE 即将提供这些能力。
最后,研究人员提醒用户在使用 FINCHES 进行预测时,需充分考虑方法的已知假设与局限。但在此前提下,FINCHES 提供了一种有效的方式,定性(乃至半定量)地洞察 IDR 与其作用对象之间的分子互作机制,为后续的突变设计和功能验证提供了可操作的研究框架。
整理 | WJM
参考资料
Ginell, G.M., Emenecker, R.J., Lotthammer, J.M., Keeley, A.T., Plassmeyer, S.P., Razo, N., Usher, E.T., Pelham, J.F. and Holehouse, A.S., 2025. Sequence-based prediction of intermolecular interactions driven by disordered regions. Science, 388(6749), p.eadq8381.
内容中包含的图片若涉及版权问题,请及时与我们联系删除


评论
沙发等你来抢