
DRUGAI
高效的新药发现依赖于候选分子的可合成性,但当前基于机器学习的反应预测方法受限于高质量数据的缺乏。研究人员构建了一个基于三组分反应的按需合成平台,可生成具有药物样性的分子。通过微型化与自动化,研究人员在3微升尺度上完成了5万个反应,涵盖193种底物,建立了迄今最大的公开反应结果数据集。借助机器学习,不仅可准确预测未知反应的结果,还评估了数据规模对模型训练的影响,为探索化学反应性的机器学习方法提供了坚实的数据基础。

精准预测有机反应结果的挑战仍未解决,主要原因在于缺乏涵盖广泛分子性质与结构多样性的、规模大且无偏的数据集。三组分或更多组分参与的分子组装过程,能够从有限的起始原料中构建大量有机分子库,非常适合用于生成实验数据,从而评估和优化反应预测的计算方法。例如,已有研究广泛应用于微型化文库构建的多组分反应(如 Ugi 缩合)就展示了此类方法的潜力。
受到这些策略的启发,研究人员此前提出了一种称为“合成发酵”的策略,即无需外源试剂,将预构建的模块组装为具有功能性与立体复杂性的低聚物。研究人员意识到,通过三组分偶联也可以实现类似方式构建更具药物样性的分子,并进一步实现微型化、高通量地从有限的起始物质中合成数十万种不同化合物。这一策略为构建一个包含数万个独特反应组合结果的大规模数据集提供了契机,为基于机器学习的反应预测方法提供了理想的训练集。
目前,使用高通量实验为特定预测任务生成定制数据集已成为常规做法,但此前的数据集总量通常仅数千个反应,且涉及的底物种类也较少。尽管已有研究在预测未知底物反应结果方面取得进展,但要在多个底物维度上实现有效外推仍是一项重大挑战。此外,当前的外推测试样本数量较少,常常限制了模型评估的可靠性。如果能构建一个包含数万条反应数据的大型数据集,将有助于验证多个领域中的未解假设,包括预测模型所需的数据量、外推能力与模型类型及数据规模之间的关系等。
在本研究中,研究人员报道了一个基于三组分组装的按需反应平台,能够在微升尺度上完成 50,000 次反应,每次使用独特的底物组合。所有反应混合物均通过液相色谱-高分辨质谱(LC-HRMS)进行分析,每种组合可检测到最多八种不同产物,最终得到一个前所未有的数据集,涵盖约 40,000 个反应组合及其特定产物。研究人员利用机器学习技术准确预测了未知反应组合和未测试底物的反应结果,并进一步分析了数据集规模对模型表现的影响,从而得出关于数据效率与训练集生成的重要结论。
研究结果
反应开发与自动化
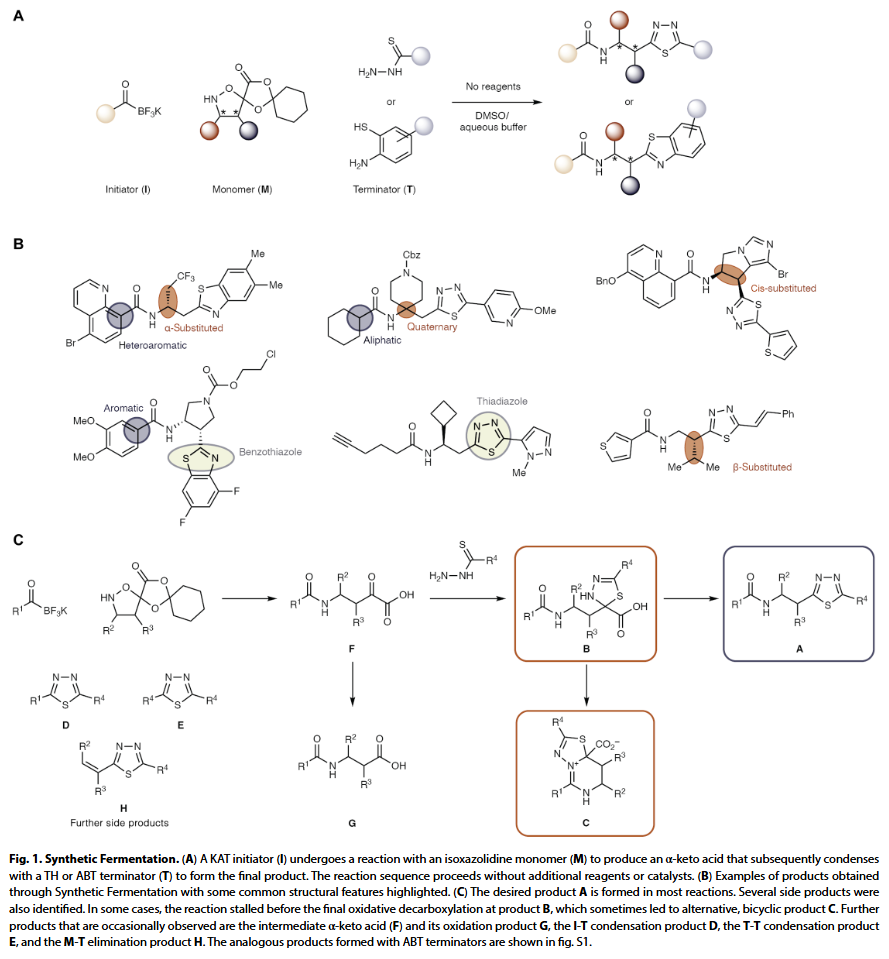
在此次“合成发酵”策略中,研究人员设计了一种三组分反应体系:以钾酰基三氟硼酸盐(KAT)为起始剂(I),与一个带有保护基团的异噁唑烷啉单体(M)反应,该保护基可释放 α-酮酸;随后,该中间体与硫代肼(TH)或氨基苯硫醇(ABT)类终止剂(T)反应生成最终产物。值得注意的是,该反应可在水性缓冲液中进行,无需催化剂或额外试剂,且副产物温和,对生物实验友好。
为了构建反应空间和训练集,研究人员共筛选了 78 种起始剂、74 种单体和 41 种终止剂。除部分用于结构多样性的特殊模块外,大多数原料易于合成,且已在此前文献中使用过。三个类别的起始剂(脂肪族、芳香族、杂芳族)、四种类型的单体(α/β-取代、顺式取代、季碳结构)以及两种类型的终止剂(ABT 和 TH)可组合生成约 236,000 个潜在产物,构成了研究人员命名的“PRIME 文库”——即这些反应可直接从已有的 193 种模块按需启动。
该三组分组装策略不仅因其产物具有良好性质而被选中,更重要的是它并非理想反应体系,因此能代表更普遍的反应预测难题。例如,虽然目标产物(A)常为主要产物,但也常出现前期中断(产物 B)、发生分子内环化(产物 C)、或形成副产物(如 I-T 缩合产物 D、终止剂二聚体 E、中间体 F、羧酸 G、或 M-T 消除产物 H)等多样性结果。这些多路径反应结果,特别是目标产物 A 与副产物 C 之间的分化,使该体系非常适合用于高通量实验与机器学习模型的验证。
为使多组分反应适用于声学分液的高通量合成流程,研究人员将反应溶剂从 t-BuOH 改为 DMSO/1 M 草酸,并将原料浓度降至 50 mM,以保证溶解性。大部分模块在 −20°C 下的 DMSO(I 和 M)或反应液(T)中可长期稳定保存。为减少原料和耗材消耗,研究人员采用声学分液技术自动配液,在 384 孔板中开展反应,每个反应体积仅为 3.3 μL,即使用 9:1 的 DMSO/草酸混合溶液。即便在如此低体积下,于 60°C 孵育期间也未观察到明显蒸发。需要说明的是,这些条件是针对高通量筛选的小规模合成而优化的,若需大规模制备单一化合物,可通过调整条件(如加入铵盐、延长反应时间、升高温度或添加氧化剂)进一步优化产率和纯度。
5 万次反应的执行与评估
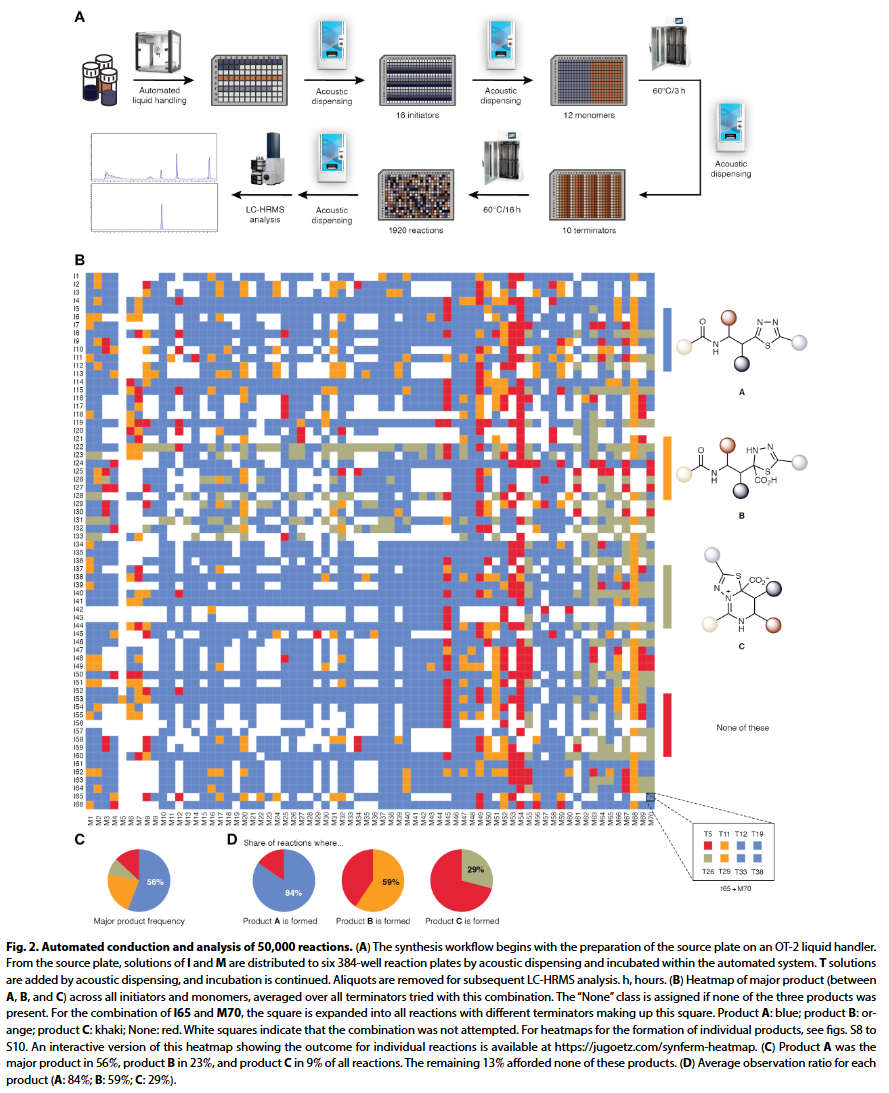
研究人员以每批 1920 次反应的规模开展文库合成,每次使用 16 种起始剂、12 种单体和 10 种终止剂,完成一批实验大约需 22 小时。通过引入自动化流程优化,例如将孵育过程集成进自动系统,并用编程化液体工作站替代手工操作,研究人员实现了每天处理两批(3840 个反应)。自动化工作流程中,原料经质控检测后加入源板,再由声学分液装置分配至六个 384 孔合成板中,起始剂与单体在 60°C 下孵育 3 小时后加入终止剂,并在同一温度下过夜反应。整个过程包括分液、孵育、转板、离心、封板等步骤均无需人工干预。
分析 5 万个反应(每个反应起始物和产物均不同)是一项挑战。研究人员使用自动系统进行样品前处理,将 30 纳升反应液稀释 1000 倍,并添加内标进行 LC-HRMS 分析。为保证分辨率,每个样品的运行时间为 14 分钟(双柱系统可缩短为 7 分钟),总分析周期约 8 个月。研究人员使用 Python/RDKit 脚本自动生成所有可能产物的分子式,并尽量减少人工干预。所有产物的峰面积均归一化至内标,通过是否检出产物给予每个反应二元标签;主要产物通过校正离子化效率后进行判定。最终数据集中,研究人员根据模块质控、分液记录和 LC-HRMS 数据等元信息进行质量筛选,剔除约 1 万条低质量数据。对 377 个重复实验的分析表明,A 类产物准确率达 98%,主产物判定的平衡准确率为 73%。最终保留约 4 万个反应结果,用于后续建模。
反应结果预测
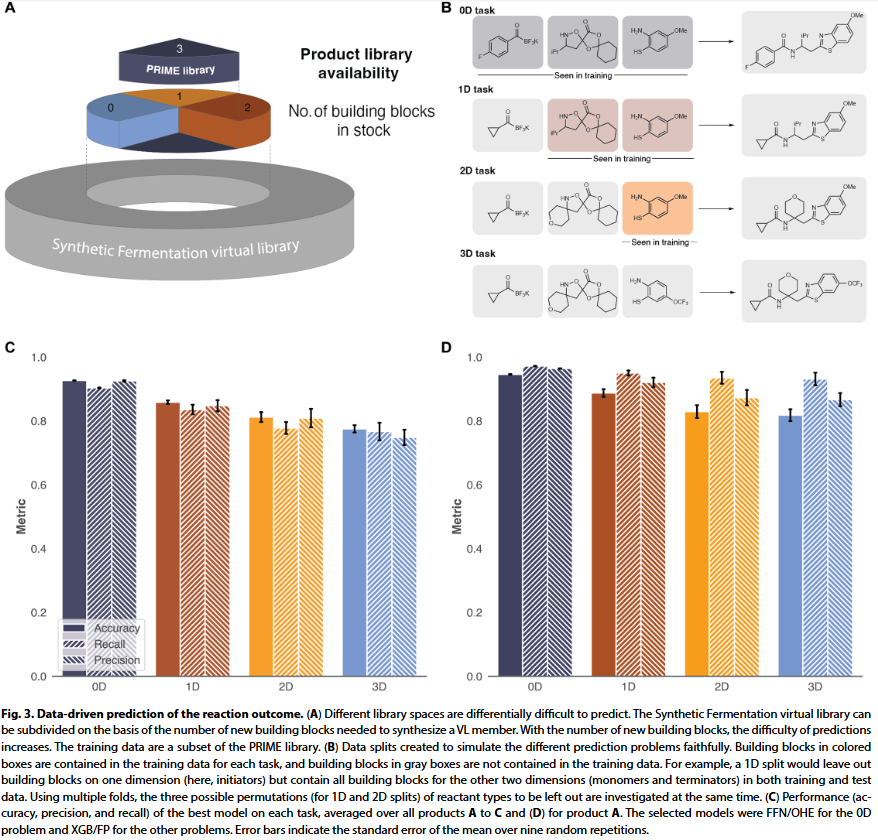
在获得大规模反应数据后,研究人员应用机器学习对任意组合的反应结果进行预测。由于为三组分反应,模型需面对多个难度等级的问题,包括训练集中是否包含 0~3 个测试反应的组分。研究人员以 A/B/C 三类产物为标签,训练多任务二分类模型,测试了结构指纹、分子属性、图表示等多种输入方式,并比较了图神经网络、前馈神经网络、梯度提升树和逻辑回归等模型。
结果显示,在训练集中包含所有组分的情形(0D 拆分),前馈神经网络配合 one-hot 编码效果最佳;而当组分缺失(1D–3D 拆分)时,梯度提升树配合结构指纹表现最优。测试集上的准确率为:0D 模型 93%、1D 86%、2D 81%、3D 78%。尽管预测结果令人满意,但研究人员指出,对于频率高的 A 类产物,仅 0D 和 1D 模型明显优于始终预测“有产物”的简单模型。随着组分维度增加,准确率下降符合预期,也说明模型需要理解真实化学信息而非仅靠组合结构。
实验验证
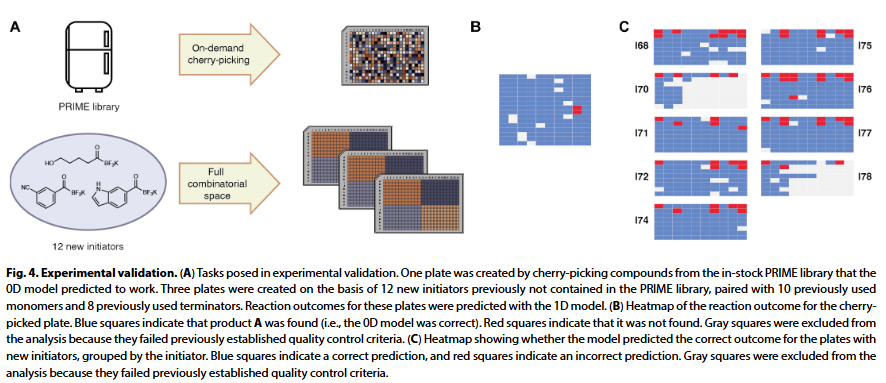
模型验证方面,研究人员进行了前瞻性测试。首先,用 0D 模型预测 PRIME 文库中可合成的反应,并实际制备其中一板化合物。结果显示,149 个预测可生成 A 类产物的反应中,147 个实验中确实生成该产物,精度达 99%。在预测 B 和 C 类产物时,精度也分别达到 87% 和 86%。
第二项测试模拟了合成优化情境:研究人员选取 12 个文库中未用过的新起始剂,结合文库中现有单体与终止剂进行反应。在 593 个有效反应中,模型成功预测了其中 533 个是否会生成 A 类产物,准确率达 90%;其中仅有 6 个为假阳性,精度达 98.9%。该结果表明模型能有效避免不必要的资源浪费,适用于先导化合物优化等实际需求。
反应预测所需的数据与模型条件
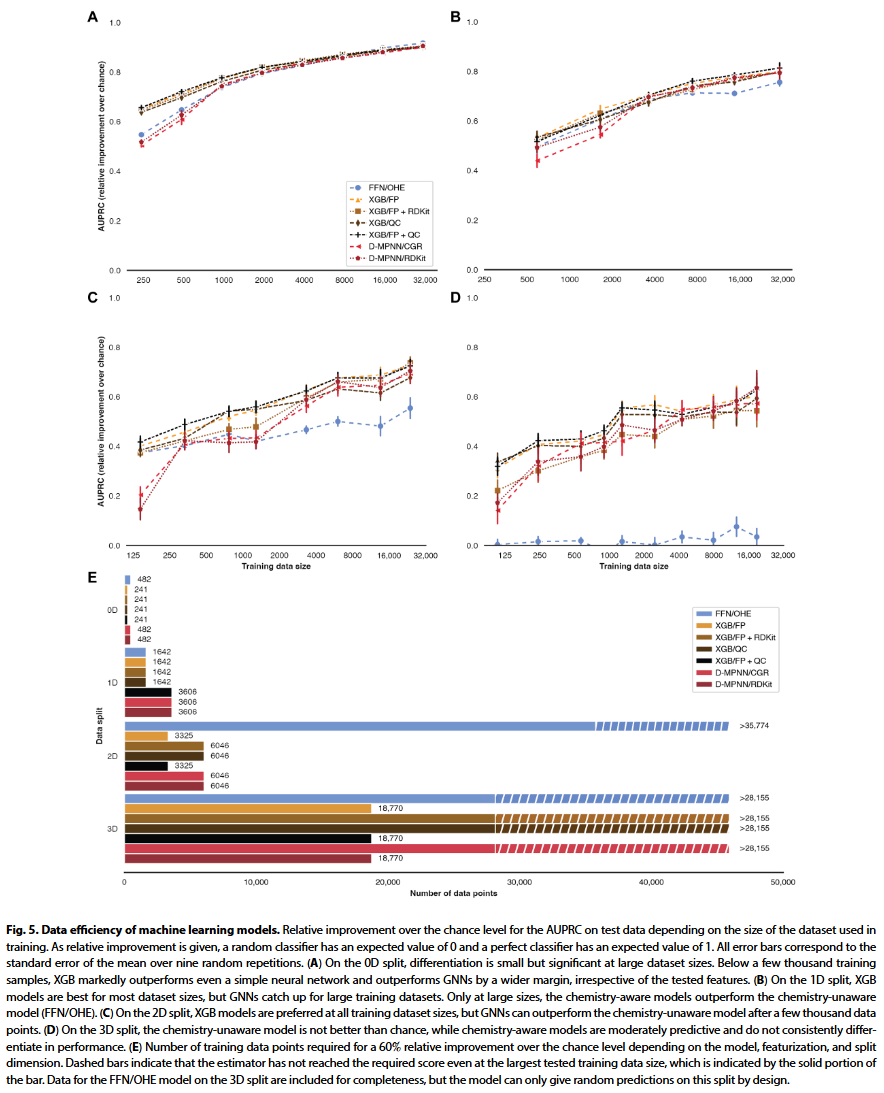
这一前所未有的大规模数据集为研究数据规模对模型性能的影响提供了独特机会。研究人员针对前述四类预测任务(0D 至 3D 拆分),分别在不同训练集规模下评估了表现最好的三类模型:FFN/OHE、XGB 和 D-MPNN。对于具有化学感知能力的模型,还引入结构指纹(XGB 使用 FP、D-MPNN 使用 CGR)或分子性质特征(RDKit)。
在 0D 拆分(纯插值任务)中,所有模型在训练数据超过 2000 条后表现趋于稳定。其中 FFN/OHE 模型在大数据量下略优于其他模型。即使训练数据量达 3 万条,模型表现仍在持续提升;而仅使用几千条数据,也能获得较好结果。在数据量小于 2000 条时,XGB 明显优于神经网络,表明其数据效率更高。1D 拆分下也展现出类似趋势,小数据集适合用 XGB,大数据量下不同模型差距逐渐缩小。值得注意的是,FFN/OHE 模型即便在数据量较大时也能表现良好,说明在一些情况下,是否具备“化学感知能力”对性能影响有限。
在更具挑战的 2D 拆分中,各模型整体表现下降,但 XGB 模型在小样本下依旧占优。FFN/OHE 模型在数据量小于几千条时表现不佳。对于最难的 3D 拆分,FFN/OHE 表现接近随机,XGB 和 D-MPNN 等模型表现虽有波动,但整体远优于随机预测。
研究人员还发现,在小数据集下引入分子性质特征(QC)可带来轻微性能提升,但在大数据量时反而可能降低效果。这种惩罚可通过将结构指纹与性质特征联合输入缓解,但提升幅度较小,不足以抵消推理过程中的计算成本。
这些结果表明,在处理组合反应预测(如 0D 拆分)时,即便仅采样约 1% 的组合空间(如 2000 条样本),也可训练出性能良好的预测模型;此时使用复杂的化学感知模型并无必要。而在 1D 外推任务中,仅当训练数据规模大于 1 万条时,化学感知模型才开始体现优势。总体而言,在当前常见的数据规模下,XGB 等简单模型往往优于更复杂的神经网络,尤其是在数据有限时。
研究人员还指出,这些结论在其他反应类型中是否适用取决于“结构-反应性”空间的复杂程度。就本研究中的合成发酵反应而言,其表现为中等“粗糙度”,大多数模块遵循简单规律(如 A 和 C 的结构趋势明显),仅少数模块存在反应性突变。因此,研究人员推测,这些发现也适用于诸如亲核取代、Suzuki 偶联等具有中等反应复杂度的体系。而对更复杂体系(如 Ni 催化反应),可能需更多数据支持预测模型。此外,该研究专注于三组分反应,但实际中许多多组分反应问题可等效转化为三维预测任务,如催化反应中的两个底物和一个配体。
总结
研究人员开发了一个自动化微量合成平台,基于三组分反应构建大量药物样化合物,目前 PRIME 文库已覆盖 220,990 个成员,起始于 188 个模块。共完成并分析了 5 万次反应,生成首个此规模的公开反应数据集。研究人员以此训练机器学习模型,实现在 PRIME 文库内的高精度预测,并展现出对新模块的良好泛化能力。模型可有效优先筛选可合成分子,并在实验中得到验证。
与此前的高通量研究相比,数据集规模的突破使研究人员能够系统评估不同模型在插值与外推任务中的需求,证实反应预测更适合作为插值问题处理,且只需稀疏采样组合空间便可获得可靠模型。在当前常见的数据量下,简单模型(如 XGB)比复杂神经网络更具优势。这些观察提示,在缺乏更高效的数据获取方式之前,建立局部、简洁、数据高效的模型是实现可靠反应预测的最优策略。复杂的模型架构和特征设计在没有新数据支持的前提下难以实现通用化的反应建模。
整理 | WJM
参考资料
Julian Götz et al. ,Predicting three-component reaction outcomes from ~40,000 miniaturized reactant combinations.
Sci. Adv.11,eadw6047(2025).DOI:10.1126/sciadv.adw6047
内容中包含的图片若涉及版权问题,请及时与我们联系删除

评论
沙发等你来抢