
DRUGAI
高亲和力的肽-人类白细胞抗原(HLA)分子结合是启动适应性免疫应答的关键。因此,精确预测肽-HLA(pHLA)结合能力对疫苗设计、肿瘤免疫治疗及自身免疫疾病机理阐明具有重要理论与应用价值。尽管基于机器学习的pHLA结合预测工具已取得显著进展,能较准确预测具有充分实验数据的HLA等位基因(约占已知HLA-I类等位基因的1%)的结合肽,但对缺乏已知结合表位报道的“零样本”(zero-shot)等位基因,其结合肽预测仍极具挑战性。此瓶颈限制了预测工具在更广人群中的应用,尤其是在需考虑个体HLA多样性的精准医疗场景。针对此难题,中国科学技术大学、加州大学圣地亚哥分校(UCSD)与南洋理工大学(NTU)的研究团队提出了一种创新的分级渐进学习(Hierarchical Progressive Learning, HPL)框架。该框架通过多层次、递进式学习策略,有效捕获并利用不同HLA等位基因间共享及特有的序列模式与结合特异性。

实验结果表明,基于HPL框架的预测模型在零样本HLA-I类等位基因上的预测性能较现有先进方法提升了60.8%;对于更罕见且研究不足的非经典HLA-I类等位基因,性能提升高达1414.0%。此外,研究团队基于高精度HPL模型,开发了自动化抗原肽突变搜索程序(Automated Peptide Mutation Search, APMS)。该程序以目标HLA等位基因和初始肽序列为输入,在HPL模型的精确指导下,通过智能化氨基酸突变策略,自动识别并优化与目标HLA具有高结合亲和力的候选肽。在超过38.1%的测试案例中,APMS仅需有限次突变,即可将弱结合或非结合肽转化为高亲和力候选肽。
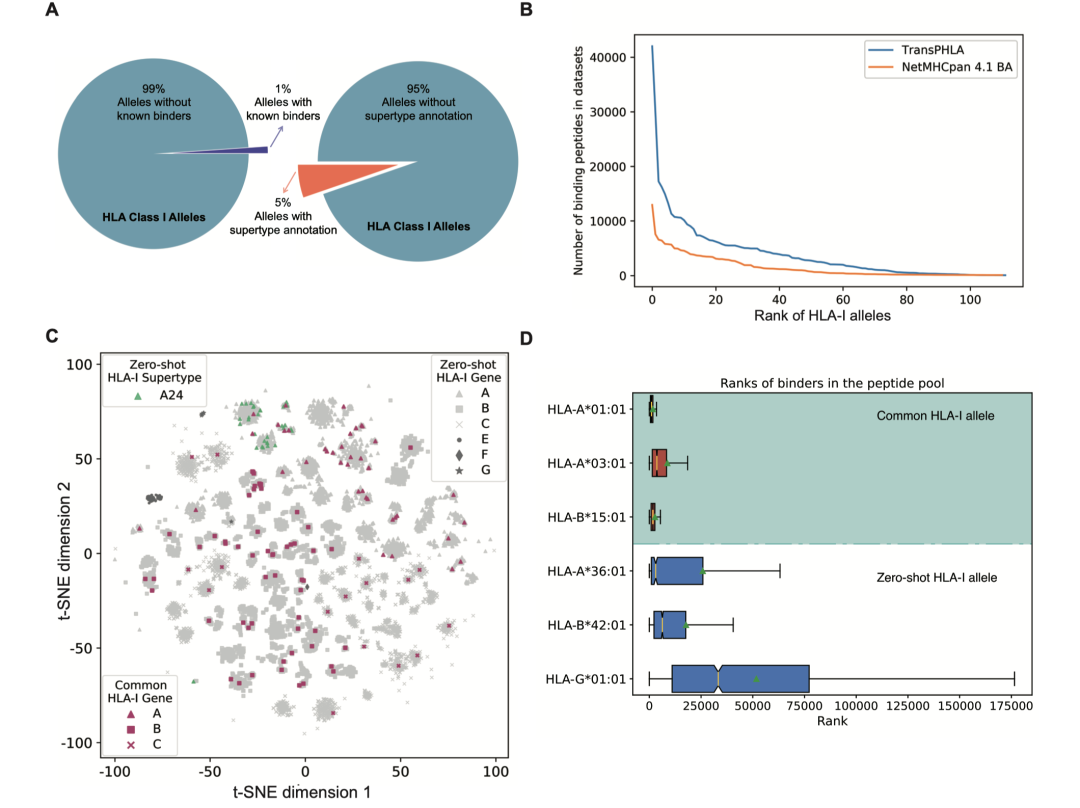
泛特异性pHLA结合预测方法在零样本HLA分型上的泛化能力瓶颈
泛特异性肽-HLA(pHLA)结合预测旨在构建适用于所有HLA分型的统一模型。然而,当应用于缺乏训练数据的“零样本”HLA分型时,现有方法的泛化能力面临严峻挑战。尽管已识别的独特蛋白质序列HLA分型超过14,000个且持续增长,但现有预测模型的训练数据仅覆盖约1%(图1A)。以TransPHLA为例,研究将其划分为训练数据中结合肽信息丰富的“常见分型”和信息匮乏的“零样本分型”。对比实验表明,TransPHLA在两者间存在显著性能差距:零样本分型的大部分测试结合肽预测排名显著偏低(图1D),表明其易将非结合肽误判为结合肽(假阳性)或遗漏潜在结合肽(假阴性),可能误导抗原肽设计。因此,提升泛特异性pHLA结合预测模型在零样本分型上的泛化能力至关重要。
为探究此泛化难题的根源,研究人员对所有已知蛋白质序列HLA分型的分布进行可视化分析。利用蛋白质语言模型提取HLA分型蛋白质序列的潜在表征,并通过t-SNE降维至二维空间(图1C)。结果显示,HLA分型依据其HLA基因(如HLA-A、HLA-B、HLA-C)聚类,表明同基因家族内分型具有高度序列和结构相似性。然而,“常见分型”在这些簇间的分布极不均衡,导致模型难以将从常见分型习得的结合模式有效迁移至缺乏代表性的“零样本分型”簇,尤其对于现有训练数据中几乎完全缺失的非经典HLA分型(如HLA-E、HLA-F、HLA-G)。超型(Supertype)注释根据HLA分型在肽结合特性上的相似性进行分组,为解决此泛化障碍提供潜在思路。图1C中特定超型(如A24超型)的小簇印证了相同超型内分型可能共享相似的肽结合特异性。然而,超型注释覆盖范围有限,当前HLA-I类超型系统仅涵盖约5%的已知HLA分型(图1A),远不足以解决大规模零样本pHLA结合预测问题,亟需探索更普适高效的知识迁移途径。为此,本研究提出的分级渐进学习(HPL)综合考量了基于生物学注释(基因、超型)、蛋白质序列相似性以及蛋白质语言模型提取的深层潜在表征等多维度信息,评估分型相似性并进行知识迁移,旨在系统性突破现有预测方法在零样本分型上的性能瓶颈。
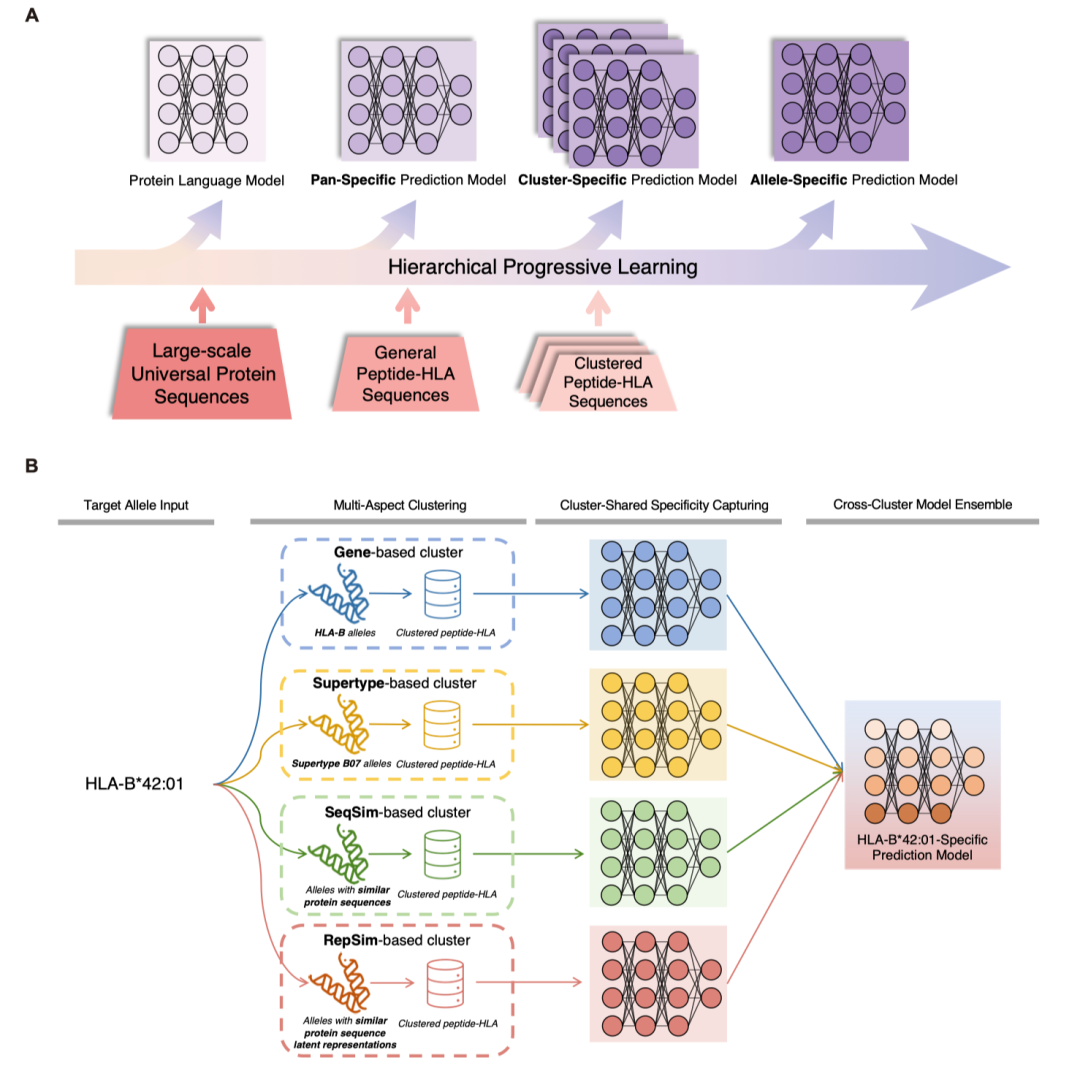
分级渐进学习(HPL)框架:实现零样本pHLA结合的精准预测
为有效应对零样本HLA分型结合肽预测的挑战,并充分挖掘分型间的关联信息,本研究创新性地构建了分级渐进学习(HPL)框架。该框架的核心在于整合了蛋白质语言模型(PLM)强大的序列理解与表征能力,以及一种定制的渐进式学习策略(图2A),以逐步识别并学习pHLA复合物序列中由普适到特异的复杂模式与关键特征。HPL旨在提供一系列在不同层级运作、且对零样本HLA分型预测能力逐步增强的pHLA结合预测模型,分别为HPL-Pan(泛特异性模型)、HPL-Cluster(簇特异性模型)和HPL-Allele(分型特异性模型)。
HPL的学习过程是一种由粗及精、逐级优化的知识提炼与迁移过程。在初始的泛特异性学习阶段,研究者利用包含多种HLA分型及其结合/非结合肽的广泛训练数据对PLM进行微调,使其在保留通用蛋白质语言知识的基础上,学习pHLA复合物序列中普遍存在的结合模式与规律(图2A)。由此训练的HPL-Pan模型作为基础模型,旨在为所有HLA分型(包括常见和零样本分型)提供初步的泛化预测能力。随后,为实现更精准的知识迁移与应用,HPL引入簇特异性学习阶段。此阶段不再等同对待所有分型,而是基于目标零样本分型的多维度特性——包括生物学注释(如HLA基因、超型)、自身蛋白质序列特征以及PLM提取的深层潜在表征——主动识别相似的HLA分型群体(分型簇)。然后,利用这些特定分型簇内的结合数据,对HPL-Pan模型进行更具针对性的微调,形成针对不同分型簇的HPL-Cluster模型(图2A, 2B)。该策略基于“物以类聚”的原则,假定簇内分型因其多方面相似性而共享更精细的肽结合模式,从而使模型能够学习到更具体和准确的结合规律。最终,为每个零样本分型构建高度定制化的最优预测模型,HPL采用分型特异性学习阶段。对于目标零样本分型,可根据其基因归属、超型信息、序列相似性等不同标准构建多个HPL-Cluster模型。通过有效整合这些来自不同相似性侧重的簇特异性模型的预测结果,最终形成该目标分型的专属HPL-Allele模型(图2B)。这种多方面聚类与跨簇模型集成的策略,旨在最大程度地捕获和利用分型间可能共享但又非完全一致的肽结合特异性,并通过综合不同视角的信息来提升预测的准确性和鲁棒性。图2B以HLA-B*42:01为例,展示了HPL框架从输入目标分型,到通过多方面聚类形成不同分型簇,再到训练簇特异性模型并最终集成形成HLA-B*42:01特异性预测模型的完整流程。通过这种分级渐进、层层深入的学习范式,HPL框架旨在逐步聚焦并精准学习由普适到高度特异的pHLA结合规律,从而有望显著提升对零样本HLA分型的预测性能。
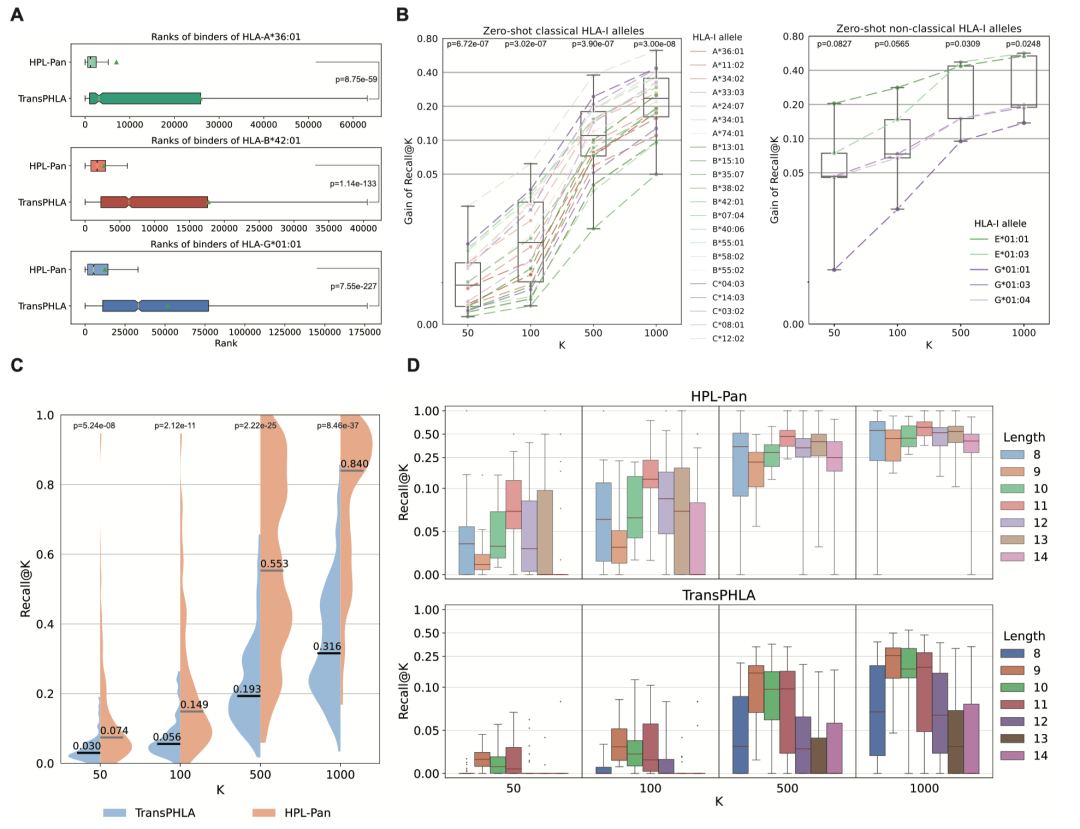
HPL-Pan模型性能评估:显著超越现有泛特异性预测方法
HPL框架的首个核心组件——HPL-Pan模型,作为基础性的泛特异性预测模型,旨在学习并理解肽-HLA(pHLA)复合物序列中普遍存在的通用结合模式。研究团队在两个专门构建的零样本测试数据集(包含经典HLA-A、-B、-C分型的“零样本经典数据集”和包含非经典HLA-E、-G分型的“零样本非经典数据集”)上,采用Recall@K作为主要评价指标。Recall@K用于衡量在预测得分排名前K的肽段中,真实结合肽的比例。如图3B所示,在所有K值设定下,HPL-Pan的Recall@K均持续优于TransPHLA,有力地证明了HPL-Pan在识别各类零样本分型(无论是经典的还是非经典的)结合肽方面的卓越能力。尤为显著的是,随着K值的增大,HPL-Pan相对于TransPHLA的性能提升呈现出扩大的趋势。考虑到优秀的泛特异性预测模型不仅应在具有挑战性的零样本分型上表现出色,在已有较多研究的常见分型上也应具备良好的预测准确性,研究团队进一步在“常见分型数据集”上对比评估了HPL-Pan与TransPHLA,结果再次证实了HPL-Pan的优势(图3C),观察到了与在零样本数据集上类似的性能提升。通过与文献报道的其他泛特异性预测方法进行直接或间接比较,也一致表明HPL-Pan在预测性能上处于领先地位(表S5)。这些全面的实验结果有力地证实,蛋白质语言模型(PLM)的引入并结合HPL框架的精心设计,能够显著提升pHLA结合预测任务的性能,使得HPL-Pan作为基于PLM的泛特异性模型,其表现显著优于TransPHLA等现有先进方法。
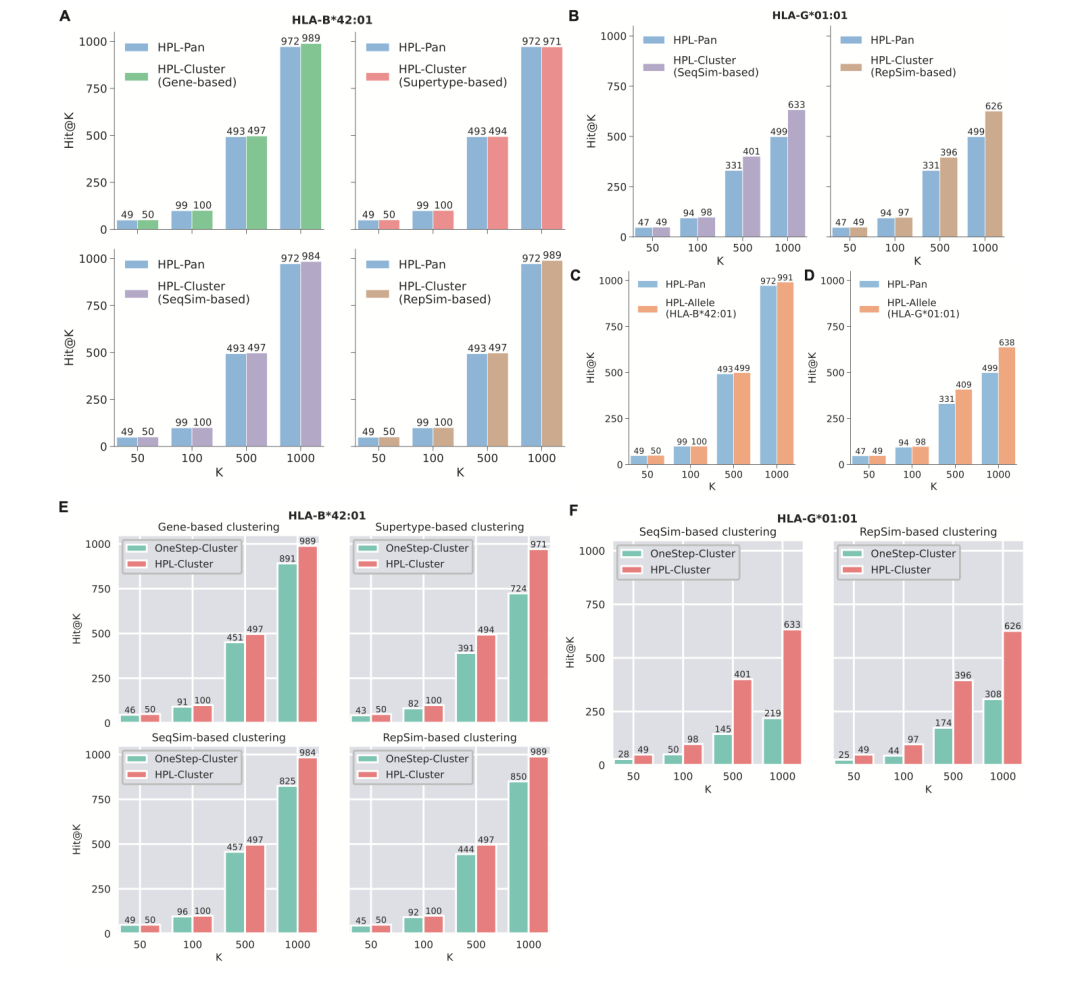
HPL-Cluster与HPL-Allele模型性能评估:渐进学习带来持续提升
在HPL-Pan模型提供的泛特异性预测基石之上,HPL框架通过簇特异性与分型特异性的渐进学习,旨在进一步提升对零样本HLA分型结合肽预测的性能。为深入探究这两个更精细层级模型的效能,研究团队选取了经典零样本HLA分型HLA-B*42:01和非经典零样本HLA分型HLA-G*01:01这两个代表性案例进行细致分析与评估。针对HLA-B*42:01,研究人员依据图2B详述的四种分型聚类策略——基于HLA基因归属、已知超型分类、氨基酸序列直接相似性(SeqSim)以及蛋白质序列潜在表征相似性(RepSim),分别构建了四个对应的HPL-Cluster模型。与基础HPL-Pan模型在识别HLA-B42:01结合肽方面的性能比较显示,所有簇特异性模型均优于HPL-Pan,其中基于基因、序列相似性和表征相似性的HPL-Cluster模型性能提升尤为显著(图4A)。这一系列性能增益验证了HPL框架通过在具有内在相似性的分型簇内进行知识共享与迁移,能够学习到更精细准确的肽结合模式,从而超越泛特异性模型表现的核心思想的合理性与有效性。更进一步,研究团队通过集成上述多个不同来源的HPL-Cluster模型的预测结果,构建了针对HLA-B*42:01的分型特异性预测模型(HPL-Allele模型)。该模型识别出的结合肽数量超过了任何单一的HPL-Cluster模型(图4C),清晰地表明整合来自不同视角(不同聚类策略)的分型簇所学习到的知识能够带来额外的性能提升。这暗示了基于生物学先验知识、序列直接比对以及序列深层语义表征所定义的分型相似性,各自蕴含关于肽结合特异性的互补信息,有效结合这些信息能够实现更优的预测效果。由于标准训练数据集中缺乏与HLA-G相关的常见分型信息,且HLA-G分型未被纳入现有超型注释体系,研究人员仅依据基因归属和表征相似性策略为HLA-G*01:01构建了两个对应的HPL-Cluster模型。尽管可用的聚类信息相对有限,实验结果显示,基础HPL-Pan模型、这两个HPL-Cluster模型以及最终集成的HPL-Allele模型在预测HLA-G01:01结合肽方面的性能依然呈现出与HLA-B*42:01案例中类似的逐步提升趋势(图4B,D,表S5C)。这一结果再次有力地证实了HPL模型(包括HPL-Cluster和HPL-Allele)在预测不同类型零样本分型(经典与非经典)结合肽方面所具有的渐进学习能力和广泛有效性。
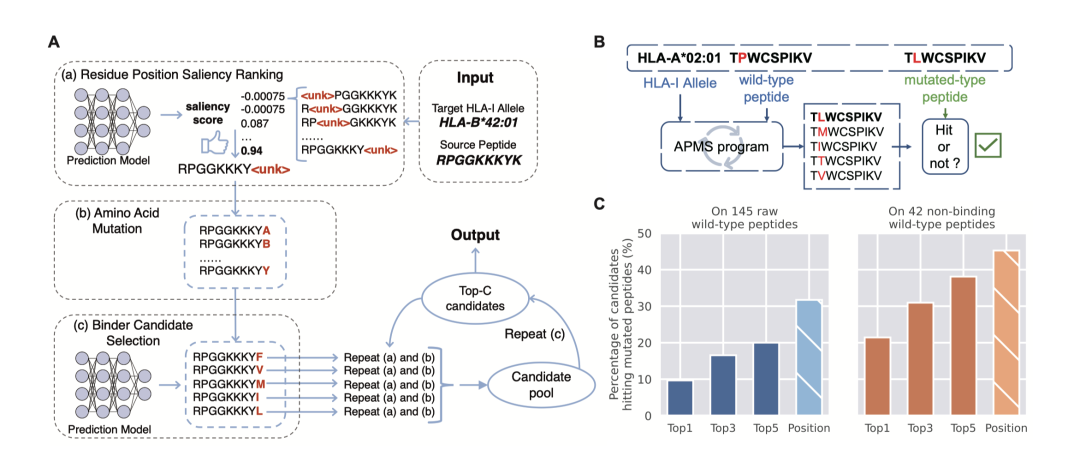
自动化肽突变搜索(APMS)程序
在获得HPL系列pHLA结合预测模型之后,研究团队进一步开发了一款名为自动化肽突变搜索(APMS)的算法,旨在辅助研究人员实现抗原肽的全自动化、高效率搜索与发现。APMS的核心优势在于其能够充分利用HPL模型提供的精确结合亲和力预测能力。当用户输入目标HLA分型以及初始肽序列(如弱结合肽、非结合肽或待优化免疫原性肽)时,APMS通过一系列精心设计的、有限轮次的迭代搜索与突变操作,自动生成一批具有较高结合潜力的候选肽序列,以供后续实验验证。
APMS的核心工作流程是一个基于预测模型反馈的迭代优化过程,包含以下三个相互关联的关键步骤(如图5A):(a)残基位置显著性排序:APMS首先评估当前候选肽序列中每个氨基酸残基位置对整体肽-HLA结合亲和力的贡献或重要性。具体方法是对肽序列的每个位置进行虚拟“扰动”(替换为“未知”氨基酸占位符),然后利用预选的HPL预测模型计算扰动前后肽序列与目标HLA分型结合分数的差异。该差异值即为该位置的“显著性分数”。通过比较所有位置的显著性分数,APMS识别出对结合影响最大的关键残基位置。(b)氨基酸突变:确定一个或多个关键残基位置后,APMS在这些位置系统性地尝试将原始氨基酸替换为所有其他19种天然氨基酸(或预定义的氨基酸替换子集),从而为每个原始候选肽生成一系列单点或多点突变的“子代”肽序列。(c)结合肽候选者筛选与迭代:所有新生成的肽序列均再次通过HPL预测模型进行结合亲和力打分。APMS根据预测得分对所有新肽序列进行排序,并从中挑选出得分最高的若干候选肽(例如Top-C个)作为下一轮迭代搜索的“种子”或直接输出为最终候选肽池。在多轮搜索模式下,APMS重复执行残基位置显著性排序和氨基酸突变步骤,持续更新和优化候选肽池,直至达到用户预设的最大搜索轮次、候选肽的预测结合亲和力达到满意阈值,或连续几轮搜索未能带来显著性能提升时终止。为系统评估APMS程序在提升肽与HLA结合亲和力方面的实际效能,研究团队构建了一个“亲和力导向突变数据集”基准测试集,并在其上对APMS进行了一轮(R=1)突变的成对评估。该数据集来源于已发表文献的实验验证数据,每条记录包含一个野生型肽、一个目标HLA分型,以及一个与野生型肽仅相差单个氨基酸残基但与目标HLA分型结合亲和力显著更高的突变型肽。实验结果显示,在总共145个野生型肽的测试案例中,当APMS仅进行一轮突变搜索时,其生成的排名第一(Top-1)的候选结合肽中,有9.7%能够精确匹配到数据集中对应的、实验验证具有更高亲和力的突变型肽。当考察范围扩展至APMS给出的Top-5候选肽时,其成功率更是跃升至38.1%(图5C)。
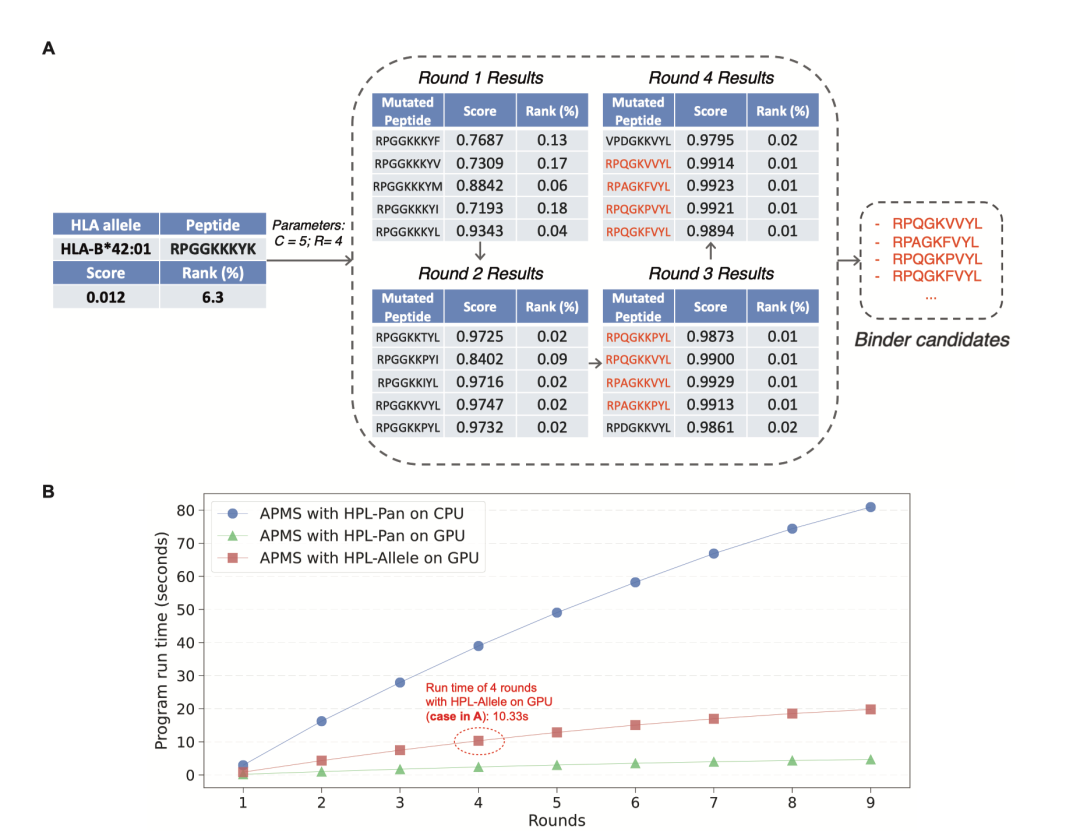
APMS(Automated Peptide Mutation Search)程序的核心优势不仅体现在其单轮突变优化肽结合亲和力的能力上,更重要的是,它能通过多轮迭代搜索机制实现对候选结合肽的持续性优化,从而探索更广阔的序列空间并发现具有更高结合潜力的肽段。图6A展示了 APMS是如何从一个已知的非结合肽出发,针对一个具有挑战性的零样本HLA分型HLA-B*42:01,逐步生成具有高度预测结合亲和力的候选肽序列的过程。随着APMS优化轮次的增加(在该案例中进行了4轮优化,每轮保留5个最优候选,即C=5, R=4),候选肽的预测结合分数呈现出显著的、持续的提升趋势:最初的肽段其NetMHCpan-4.1预测得分仅为0.012,而在经过APMS四轮的迭代优化之后,最优候选肽的预测得分已升至0.99,显示出极强的预测结合能力。更值得注意的是,在最终生成的优化肽库中,有八个候选肽在与初始源肽仅存在四个氨基酸差异的情况下,其NetMHCpan-4.1预测的百分位排名均达到了0.01%的极高水平。这一结果充分证实了APMS在为信息匮乏的零样本分型高效生成具有潜在强结合能力的候选肽方面的有效性。
总结与展望
HPL框架的核心优势之一在于它能够提供一套在三个不同粒度级别上运作的、相互关联且性能逐级提升的分层预测模型,即:泛特异性(pan-specific)模型、簇特异性(cluster-specific)模型,以及分型特异性(allele-specific)模型。HPL系列模型所展现出的、能够在信息匮乏的零样本分型上实现有效泛化的能力,对于成功应对人类HLA分型所具有的巨大多样性(polymorphism)而言至关重要,它为将来能够根据每一位患者独特的HLA基因型图谱来量身定制更为精准和有效的治疗方案提供了可能。此外,通过准确预测不同HLA分型与特定疾病相关抗原(如肿瘤新抗原、病毒抗原等)的结合情况,研究人员将能够更有效地筛选和鉴定出那些最具有治疗潜力的肽段,从而有望在更广泛的疾病类型和不同遗传背景的人群中,更大概率地诱导出强大而持久的免疫应答。
基于HPL系列预测模型,本研究提出的APMS程序能够通过一种高效的、迭代式的氨基酸突变与筛选过程,自动化地生成与目标HLA分型具有高度结合亲和力的候选肽序列。通过将APMS这样的计算工具整合到未来的免疫学研究和药物开发的实验工作流程中,研究人员将能够以更低的成本获得肽候选分子库。随后,他们便可以将主要精力集中在对这些最有前景的候选结合肽进行更具针对性的湿实验验证和功能分析上,并通过最少的实验投入来迭代地优化和确认预测出的肽候选者的实际生物学活性。
参考资料
Zhu, X., Lu, J., Hu, X., Jin, T., Lu, S., & Feng, F. (2025). Hierarchical progressive learning for zero-shot peptide-HLA binding prediction and automated antigenic peptide design. Cell Reports, 44(6).
内容中包含的图片若涉及版权问题,请及时与我们联系删除

评论
沙发等你来抢