
DRUGAI
识别能够准确捕捉蛋白质构象变化最慢时间尺度的集体变量(CV)对理解众多生物过程至关重要。研究人员提出了基于记忆核最小化的神经网络(MEMnets),该深度学习框架可精准识别生物分子动力学中的慢速CV。与常假设动力学为马尔可夫过程的方法不同,MEMnets基于积分广义主方程理论,将非马尔可夫性编码为CV的连续记忆核中。MEMnets的核心创新在于,通过并行编码器网络最小化时间积分记忆核的上界,以识别最优CV。研究人员展示了MEMnets可有效识别FIP35 WW结构域折叠过程中的慢速CV,揭示了两个并行的折叠通路。此外,通过将其应用于更复杂的细菌RNA聚合酶钳结构打开过程,进一步展示了MEMnets在采样有限的情况下依然具有稳健的数值表现。

探索生物分子构象变化有助于理解多种关键生物过程的分子机制,包括蛋白质折叠、蛋白–配体结合以及构象状态之间的转换。分子动力学(MD)模拟是一种强有力的工具,常用于补充实验手段以解析这些动态过程。然而,由于显式溶剂模型涉及大量原子,MD模拟数据本质上具有高维特征,因此仅通过分析这些数据难以获取清晰的物理图像。
构象变化往往伴随着多个显著慢速的时间尺度。将这些慢速动态模式与快速模式区分开来,有助于用少量CV揭示功能相关的构象变化。对于简单系统,可以根据物理直觉选择CV(如RMSD、扭转角、原子间距离等),但在更复杂系统中,这些CV往往难以明确选择,且缺乏物理或生物学解释性。
识别能够捕捉系统最慢动力学的CV对理解分子动力学至关重要。尽管已有方法(如主成分分析、扩散映射、ISOMAP等)试图从几何角度提取CV,但它们通常难以应对复杂的生物分子体系,因为几何变化不一定与慢速动力学相关。
基于变分原理的方法如tICA、VAMPnets和SRVs,能够识别多个慢速动态模式,但这类方法依赖于马尔可夫假设。而一旦将动力学投影至低维空间,就会引入记忆效应,从而变得非马尔可夫,尤其是在时间尺度未能充分分离的情况下。此外,这些方法在构象采样不足时容易训练失败或过拟合。
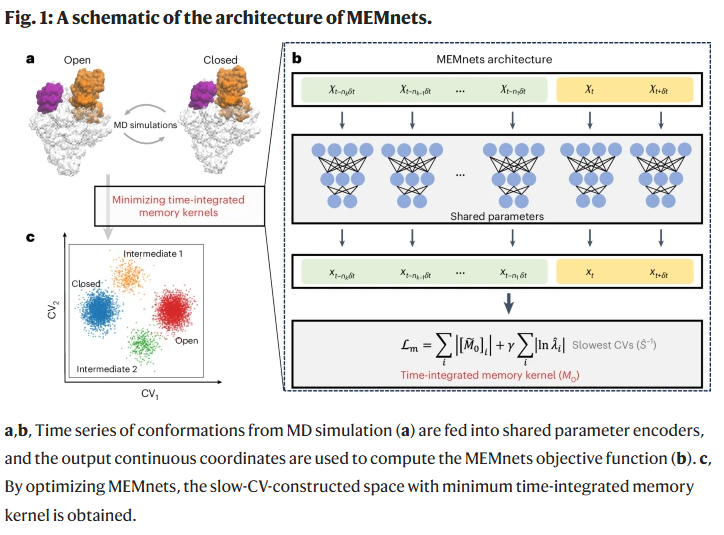
结果
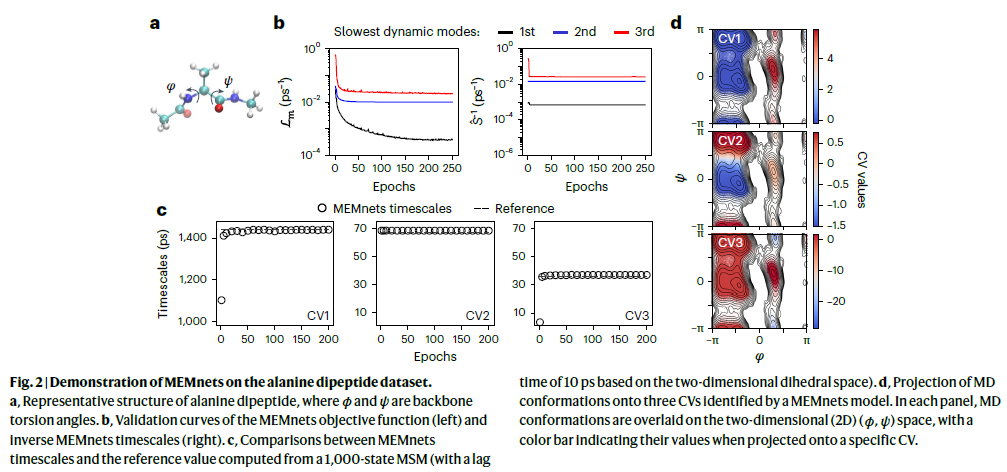
利用MEMnets最小化丙氨酸二肽的记忆核
研究人员以丙氨酸二肽为示例,展示了如何在MEMnets中优化时间积分记忆核。训练中使用了10个重原子的30个笛卡尔坐标作为输入特征,目标是提取三个集体变量(CVs)。选取的编码器网络使用了7、8、9和10皮秒的滞后时间来计算目标函数中的项。此前已有研究表明该体系的记忆核弛豫时间约为1.5皮秒,因此所选滞后时间已超过该时间尺度。
训练200轮之后,所有三个CV的损失函数值趋于稳定,表明已得到具有最小记忆的最优CV投影。进一步计算出的时间尺度表明,模型能迅速捕捉该体系最慢的三个动态模式。通过与基于1,000状态马尔可夫模型的参考时间尺度比较,结果显示一致性极佳。此外,研究人员将识别出的三个CV分别投影至由扭转角ψ与ϕ构成的二维空间中,清晰地展示了各个CV所代表的从一个自由能低谷向另一个低谷过渡的构象转变。
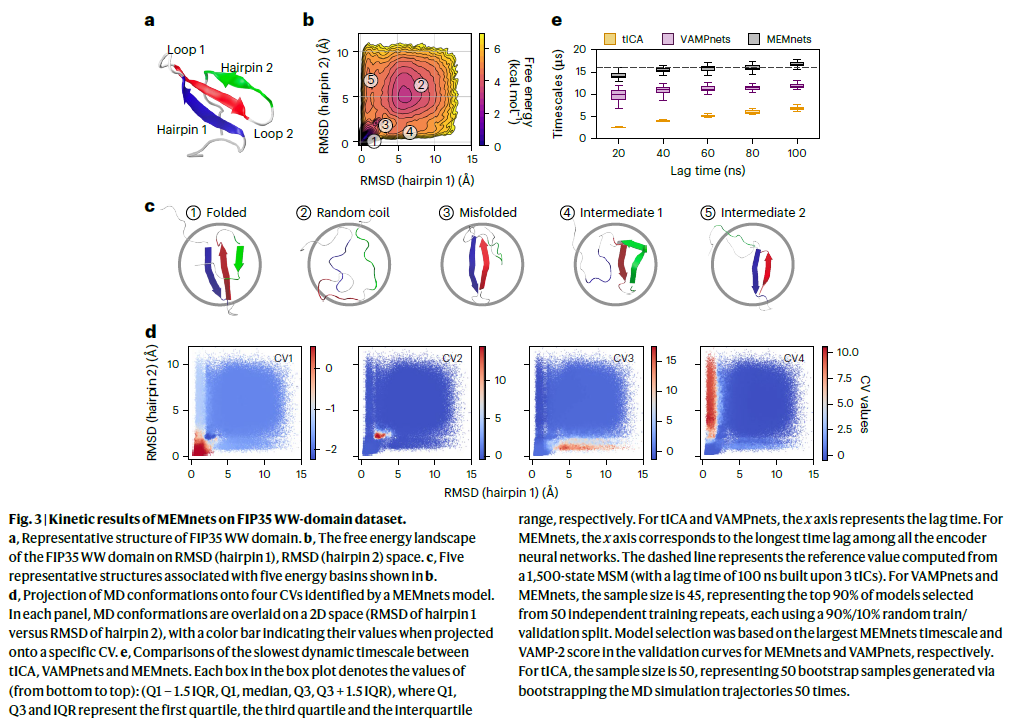
FIP35 WW结构域的折叠动力学
为了识别FIP35 WW结构域折叠过程中的CV,研究人员使用35个Cα原子之间的595对距离作为输入特征进行训练。训练在50轮内收敛,并识别出4个CV,其对应的时间积分记忆核最小。所得CV成功描述了该结构域的折叠过程,并揭示出两个截然不同的折叠路径。
为进一步分析CV与折叠路径的关系,研究人员将4个CV投影至由发夹1与发夹2相对晶体结构的RMSD定义的二维空间中,颜色从蓝到红表示CV的数值变化。投影所得的自由能景观展示了不同的自由能盆,并对每一盆提供了代表性结构。
第一个(最慢的)CV揭示了从未折叠态向折叠态的转变过程。第二个CV捕捉到一个非主通道的错误折叠中间态的跃迁。第三和第四个CV则分别对应两条平行的折叠路径:一条路径中发夹2先折叠,另一条路径中发夹1先折叠。这些路径与先前基于马尔可夫模型的研究结果一致,提供了更连续且高分辨率的视角。
此外,研究人员基于第一个CV量化了整体折叠时间,定义CV1 ≥ 0.888为未折叠态,CV1 < 0.888为折叠态。得到的平均首达时间为14.0±5.8微秒,与同一轨迹下D.E. Shaw实验室的模拟值(10±3微秒)及实验测量值(约14微秒)一致。
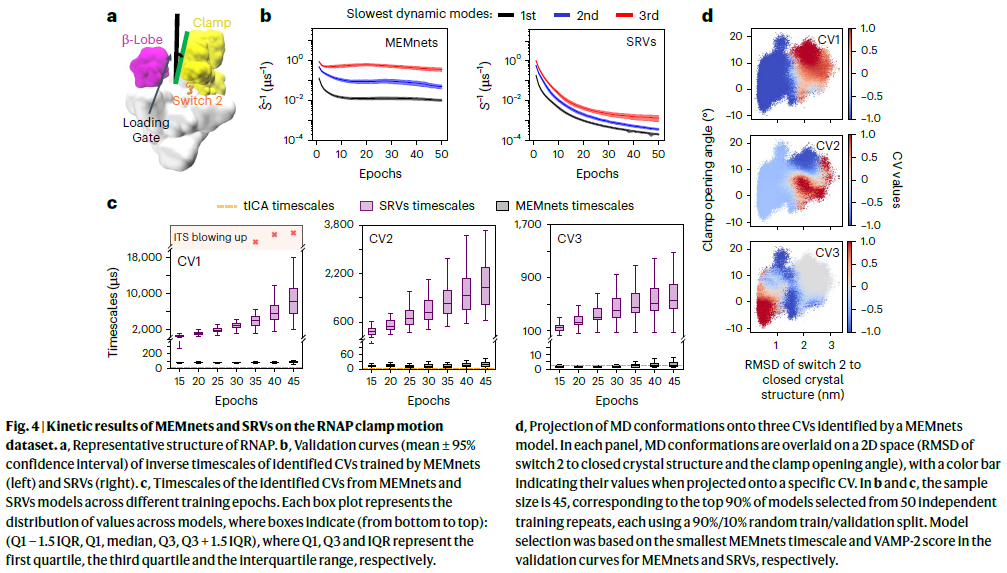
细菌RNA聚合酶的钳结构运动
研究人员进一步验证了MEMnets在更复杂系统中的表现,将其用于分析嗜热菌RNA聚合酶(Taq RNAP)中加载门的打开动力学。该加载门由钳结构与β-lobe结构构成,在转录启动时需发生大幅张开。为训练模型,研究人员使用306条200纳秒的MD轨迹,并提取1,770对原子距离作为输入特征。
考虑到此前研究中估算的记忆核弛豫时间为25~30纳秒,训练时选用了25至50纳秒范围内的滞后时间。训练结果显示,模型成功识别出三个CV,其训练过程表现出良好的收敛性。
投影分析进一步表明,CV1与switch 2区域的构象变化密切相关,该区域的折叠状态决定了钳结构的张开程度。CV2与CV3则同时关联了switch 2与钳结构本体的变化,说明这两部分之间存在协同运动。总体而言,这三个CV揭示了Taq RNAP加载门开启的分子机制,即switch 2区域作为铰链引导钳结构张开。
基于MEMnets识别的CV构建RNA聚合酶的状态模型
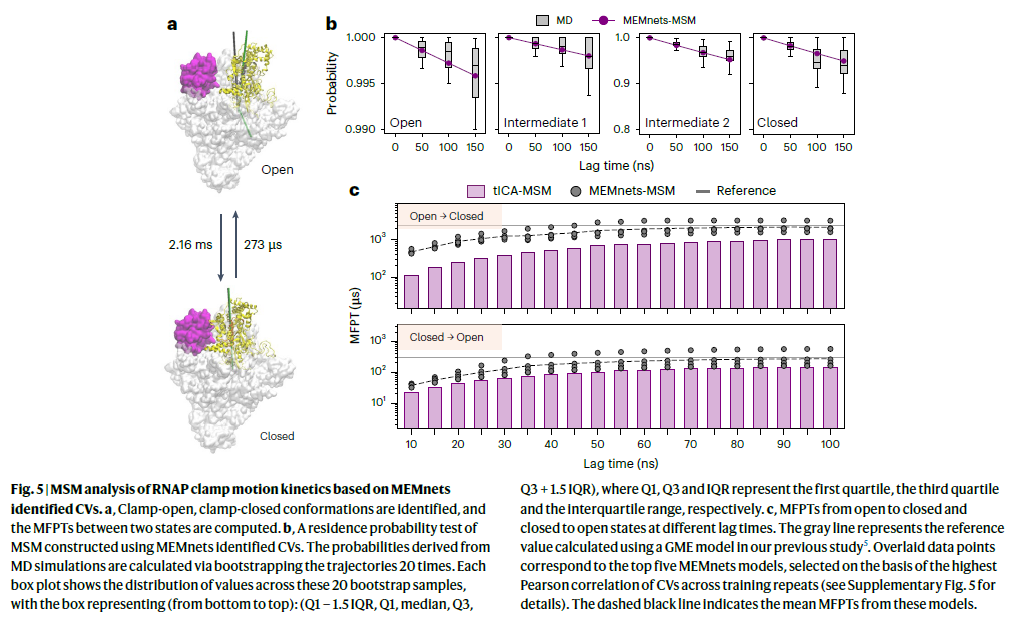
研究人员进一步评估了MEMnets在构象聚类与状态建模中的降维能力。在此基础上,研究人员将所有MD构象投影至MEMnets识别出的三个CV,并通过K-means聚类将其划分为四个状态。这四个状态分别对应于RNA聚合酶的“钳打开”状态、“钳关闭”状态及两个中间状态。
为了验证这一四状态模型的合理性,研究人员进行了Chapman–Kolmogorov测试。结果显示,该模型预测的状态驻留概率与从MD轨迹直接计算的概率高度一致,表明MEMnets提供的低维CV空间具有最小记忆性,并足以构建近似马尔可夫的有效状态模型。
研究人员进一步计算了从“打开”态至“关闭”态,以及反向路径的平均首达时间(MFPT),并与基于tICA识别CV所构建的MSM进行了对比。结果显示,MEMnets-MSM的MFPT大约是tICA-MSM的两倍,且与以往研究中基于GME模型得到的MFPT相符,进一步强调了MEMnets在构建记忆最小化的动力学投影空间方面的优势。
MEMnets在训练收敛性方面优于SRVs
在复杂生物体系中,由于采样有限,SRVs与VAMPnets在训练过程中的收敛性常常成为瓶颈,因此通常需要额外设置早停准则以避免过拟合。研究人员以RNA聚合酶为例,将MEMnets与SRVs在训练过程中的表现进行了比较。
结果表明,MEMnets在训练初期即展现出稳定的验证曲线,且三组CV所对应的时间尺度均保持收敛。而SRVs的验证曲线未能收敛,时间尺度线性飙升,导致模型出现极大甚至无穷大的时间尺度,表明其陷入过拟合,捕捉到断裂而非真实的慢速动力学。
进一步地,研究人员将训练不同轮次下的SRVs模型所识别的CV投影至构象空间中,发现其结果确实表现为动力学断裂。同时,即便调整SRVs的滞后时间,也无法缓解该过拟合现象。相比之下,MEMnets通过对完整时间序列进行建模,显著增强了训练的稳定性与物理可信度。
此外,MEMnets的目标函数设计中引入了IGME理论的上界形式,进一步提升了模型的稳定性,避免出现断裂动力学的情形。
讨论
MEMnets的核心创新在于其神经网络架构能够在考虑记忆效应的同时,非线性地整合输入特征,从而跳出马尔可夫假设的限制,识别连续、最小记忆的CV。相比于依赖变分原理的tICA与VAMPnets,MEMnets不依赖于全马尔可夫动力学,可在较短的MD轨迹上仍准确建模。尽管MEMnets训练成本更高(通常因使用多个编码器网络而训练速度变慢),但其在构象变化复杂或时间尺度不完全分离的体系中尤具优势。
研究人员进一步指出,MEMnets的潜力不限于CV识别。将其扩展为在潜在空间中构建非马尔可夫动力学模型(例如基于GME理论的生成模型)具有前景。此外,未来可引入SoftMax层将其输出直接映射至离散状态,并扩展至非平衡动力学过程的建模。尽管目前基于欧几里得空间表示自由能景观,其他几何拓扑如球面潜空间的引入也可能提升其表达能力。
总之,MEMnets有望成为研究生物分子慢速动力学的重要工具。
整理 | WJM
参考资料
Liu, B., Cao, S., Boysen, J.G. et al. Memory kernel minimization-based neural networks for discovering slow collective variables of biomolecular dynamics. Nat Comput Sci (2025).
https://doi.org/10.1038/s43588-025-00815-8
内容中包含的图片若涉及版权问题,请及时与我们联系删除

评论
沙发等你来抢