
DRUGAI
逆蛋白质折叠旨在生成可折叠为目标蛋白质结构的有效氨基酸序列。尽管近年来深度学习方法在该领域表现出强大潜力,但在预测具有高度结构不确定性区域(如无序区域)方面仍存在挑战。为此,研究人员提出了一种掩码先验引导的去噪扩散(MapDiff)框架,能更准确地捕捉结构信息和残基互作。MapDiff 是一种离散扩散概率模型,可在给定蛋白骨架的条件下,逐步生成噪声降低的氨基酸序列。为融合结构与互作信息,研究人员设计了一种图神经网络,并通过掩码先验预训练提升模型能力。此外,在生成过程中,结合了去噪扩散隐式模型与蒙特卡洛 dropout,以降低不确定性。在四个序列设计基准数据集上的评估结果表明,MapDiff 显著优于现有方法,其生成的序列在理化性质和结构特征上也与天然蛋白高度一致。

蛋白质是由线性氨基酸序列折叠形成的复杂三维结构,几乎参与所有关键的生物过程,如代谢调控、免疫反应和细胞周期控制。逆蛋白质折叠(inverse protein folding, IPF)任务旨在根据指定的蛋白质三维骨架,设计出能够稳定折叠为该结构的氨基酸序列,是蛋白质设计与合成生物学中的核心问题之一。该任务的应用前景广泛,涵盖新型酶设计、抗体优化、疫苗开发和合成蛋白质工程等领域。
传统的基于物理建模的序列设计方法通常将该问题形式化为能量最小化问题,但往往面临计算开销大、建模精度有限的问题。近年来,深度学习方法逐渐成为该领域的主流手段,其具备从数据中自适应学习复杂非线性关系的能力,能有效捕捉蛋白质结构与序列之间的高阶依赖。
尽管如此,现有深度学习模型在处理结构不明确区域时仍存在显著不足,特别是对于无序区、弯曲区或灵活区的残基预测不稳定。此外,许多方法采用自回归生成方式,在长序列生成中容易积累误差,难以捕捉全局结构依赖。非自回归、迭代生成方法逐渐受到关注,显示出更优的建模能力和鲁棒性。因此,研究人员探讨了一种结构感知、可迭代去噪的生成机制,以实现更高质量的序列设计。
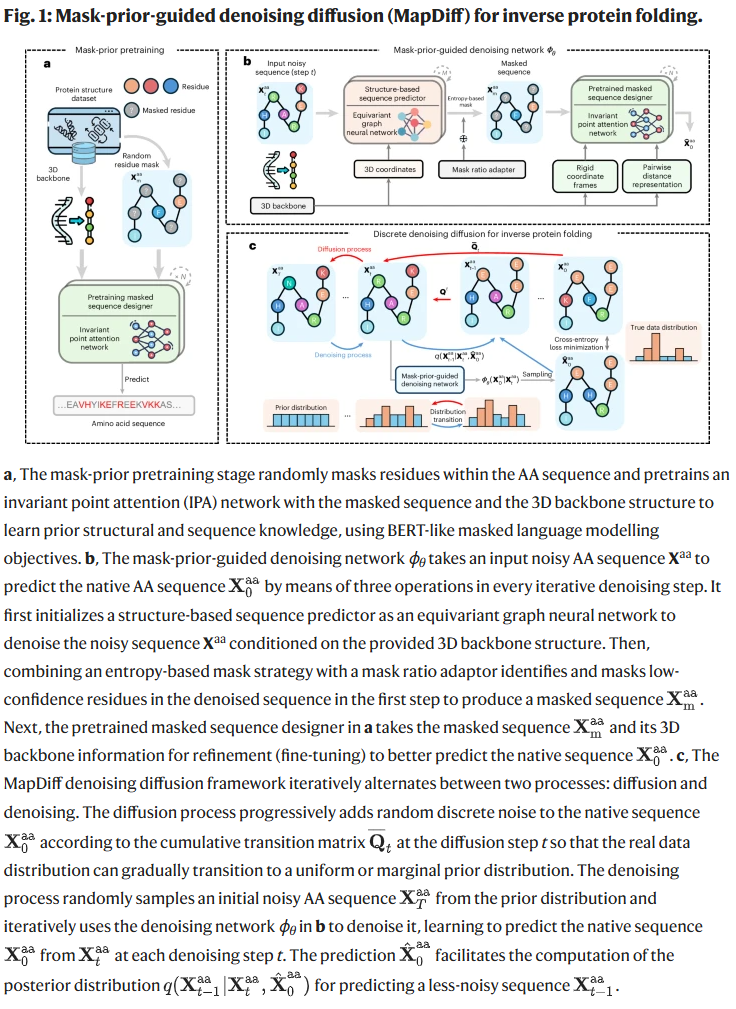
MapDiff 框架设计与基本原理
MapDiff 将逆蛋白质折叠建模为一个离散的扩散与去噪过程。通过一系列的离散噪声添加操作,原始氨基酸序列被逐渐转化为无序表示。接着,基于图神经网络的去噪网络在三维结构信息的引导下逐步还原序列。
其核心去噪网络包含三个模块:
结构感知序列预测器:利用等变图神经网络,建模残基在三维空间中的相互作用。
掩码先验机制:识别当前步骤中置信度低的残基并进行掩码处理。
预训练的掩码序列设计器:利用掩码语言建模目标,对被掩盖的残基进行重构与优化。
此外,MapDiff 引入非自回归生成策略,避免预测误差累积;结合离散版 DDIM 以跳步加速生成过程,并通过蒙特卡洛 dropout 提高序列预测置信度。
蛋白质序列恢复能力评估
研究人员在 CATH 4.2 和 CATH 4.3 两个标准蛋白质结构分类数据集上进行了系统评估,并在短序列、单链序列子集中做了进一步分析。结果表明:
MapDiff 在困惑度与残基恢复率两个核心指标上均取得最佳结果,显著优于包括 ProteinMPNN、PiFold、LM-Design、GRADE-IF 等多个先进方法;
即便在无额外知识的情况下,MapDiff 依然展现出更强的泛化能力;
无论采用均匀先验还是边际先验,模型表现稳定,表明其生成策略具有鲁棒性;
在氨基酸相似性评估中,MapDiff 的预测结果与 BLOSUM 替代矩阵高度相关,能够捕捉生物上合理的保守替换。
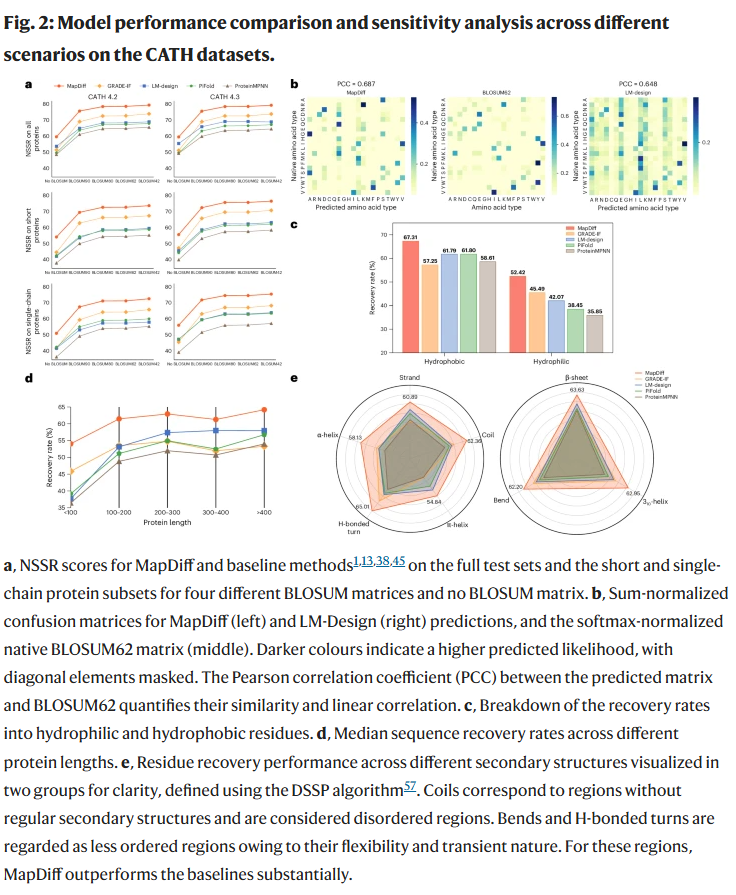
多场景泛化能力与折叠验证
在 TS50 和 PDB2022 这两个与训练数据完全独立的数据集上,MapDiff 依旧展现出领先的性能,尤其在氨基酸保守替代性指标(如 NSSR90)上显著优于其他模型,表明其具备出色的零样本迁移能力。
研究人员进一步使用 AlphaFold2 对生成的序列进行结构重构,结果显示,MapDiff 所生成的序列即使在总体序列相似度较低的情况下,也能折叠出与真实结构高度一致的三维构象,显示出优秀的可折叠性。
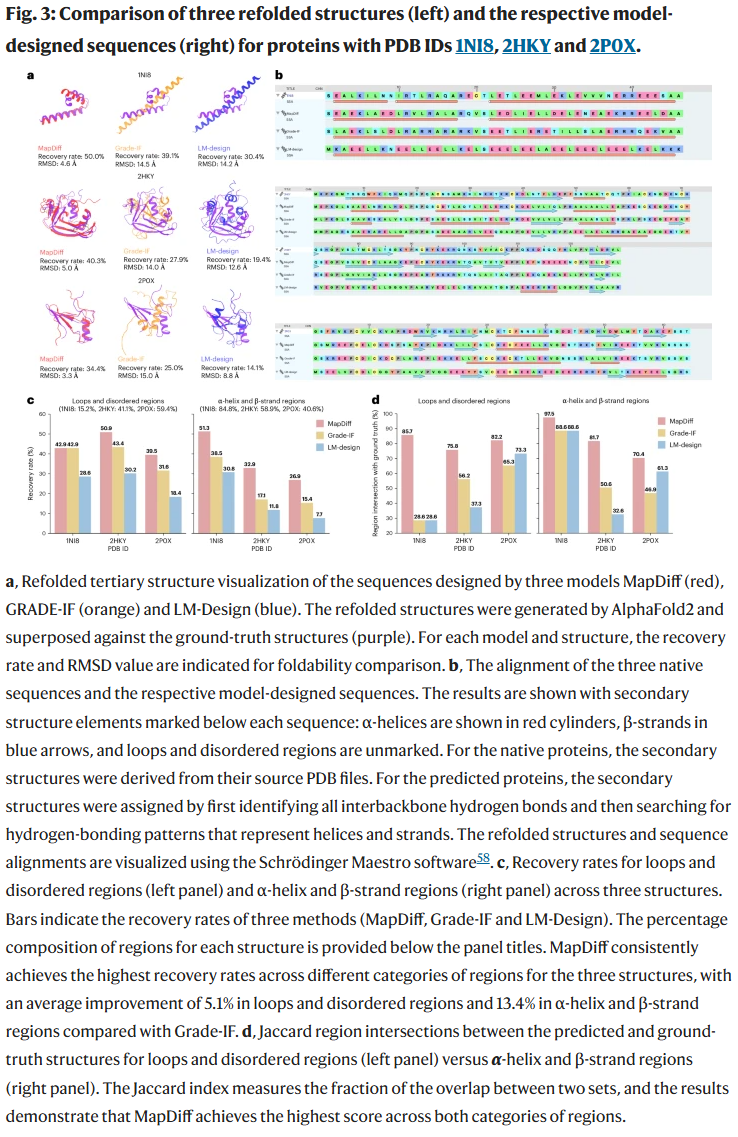
结构一致性与功能残基恢复
通过对三个代表性蛋白质(PDB ID: 1NI8, 2HKY, 2P0X)的详细比较,MapDiff 在恢复关键二级结构(如 α-螺旋、β-折叠)、识别功能残基(如正电荷残基或二硫键)方面表现更优。其所生成序列不仅结构更接近实验解析结构,在 RMSD 等指标上也显著优于对比方法。
例如在 H-NS 蛋白(1NI8)案例中,MapDiff 成功恢复关键 DNA 结合残基位置与二级结构;在人核糖核酸酶 RNase7(2HKY)中,MapDiff 预测出了 3 个正确位置的半胱氨酸并构成二硫键,而对比方法完全缺失这些关键残基。
模型消融与组件贡献分析
研究人员系统性移除 MapDiff 各模块后发现:
去除 IPA 模块导致恢复率下降超过 4%,表明其在低置信残基优化中作用显著;
去除全局上下文或坐标更新机制,也显著削弱模型预测能力;
各子模块对折叠性指标影响略低于对序列恢复的影响,说明 AlphaFold2 在结构预测中具有一定的容错能力。
讨论
研究人员提出的 MapDiff 框架突破了现有逆折叠方法在建模复杂序列-结构映射中的局限,展示了掩码引导扩散模型在蛋白质设计任务中的巨大潜力。MapDiff 通过引入掩码先验预训练策略与结构引导的图神经网络,有效融合序列上下文与三维空间结构信息;而通过 DDIM 与 dropout 技术,进一步提升了生成效率与鲁棒性。
实验结果表明,MapDiff 不仅在传统指标上优于现有方法,且能生成具有实际折叠能力的序列,即便训练数据受限亦能保持较强的泛化能力。未来研究可进一步探索 MapDiff 在抗体设计、酶催化位点优化、结合口袋特异性设计等任务中的应用前景,并结合蛋白语言模型、结构预测工具或物理约束机制增强其生物学可解释性与实验实用性。同时,开展结构模拟或分子动力学验证将有助于进一步检验其生成序列的稳定性与功能潜力。
整理 | WJM
参考资料
Bai, P., Miljković, F., Liu, X. et al. Mask-prior-guided denoising diffusion improves inverse protein folding. Nat Mach Intell (2025).
https://doi.org/10.1038/s42256-025-01042-6
内容中包含的图片若涉及版权问题,请及时与我们联系删除

评论
沙发等你来抢