
DRUGAI
2025年6月11日,大连理工大学以《DeePEST-OS: A Generic Machine Learning Potential for Accelerating Transition State Search in Organic Synthesis》为题,在ChemRxiv(https://chemrxiv.org/engage/chemrxiv/article-details/684161351a8f9bdab5d606ae)上预发表了机器学习势函数领域的最新研究进展。
在有机合成领域,精准解析过渡态结构与反应势垒是理解反应动力学的关键,但传统密度泛函理论(Density Functional Theory, DFT)计算成本高、耗时长,限制了大规模反应机制研究。对此,本研究提出了一种适用于有机合成的通用反应机器学习势函数模型——DeePEST-OS (Deep learning-based molecular Potential Energy Surface prediction Tool for Organic Synthesis),建模方法如图1所示。该模型基于高精度ωB97M-V/def2-TZVP计算方法,可实现涵盖10种化学元素(C、H、O、N、P、S、F、Cl、Br、I)的有机合成过渡态搜索,在保持DFT计算精度的情况下(能量MAE=0.266 kcal/mol、力MAE=0.380 kcal/(mol·Å)),计算速度提高超1000倍。此外,DeePEST-OS模型可评估过渡态构象异构体的能量大小,并可集成逆合成规划算法,可实现高效精准预测药物逆合成路线的反应势能面的目标。
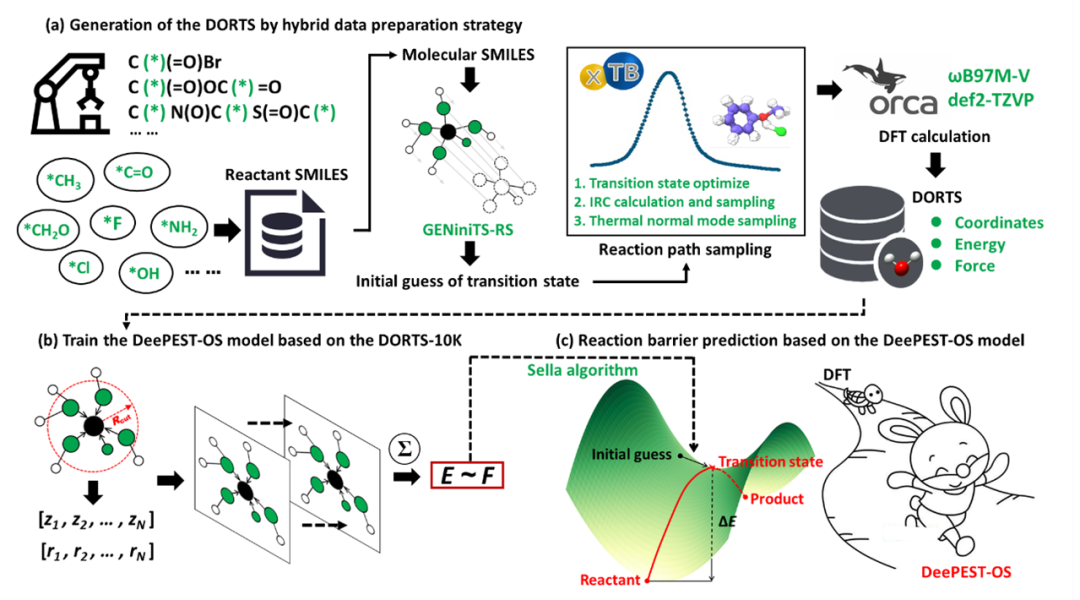
图 1. DeePEST-OS建模方法
全新海量反应数据库
DeePEST-OS模型基于一个全新海量反应数据库DORTS(Database of Organic Reaction Transition States)进行训练。与目前主流的Transition1x反应数据库相比,DORTS数据库在化学元素种类、反应类型、反应数量和DFT计算水平上都实现了突破,如表1所示。DORTS中的反应来源于有机化学经典教材《March高等有机化学》,涵盖了有机合成领域常见的化学反应。反应过程中的分子构象经过渡态搜索、IRC(Intrinsic Reaction Coordinate)计算、简正模采样获得。DORTS的化学空间基本覆盖了Transition1x,如图2所示。
表1 DORTS数据库对比Transition1x数据库
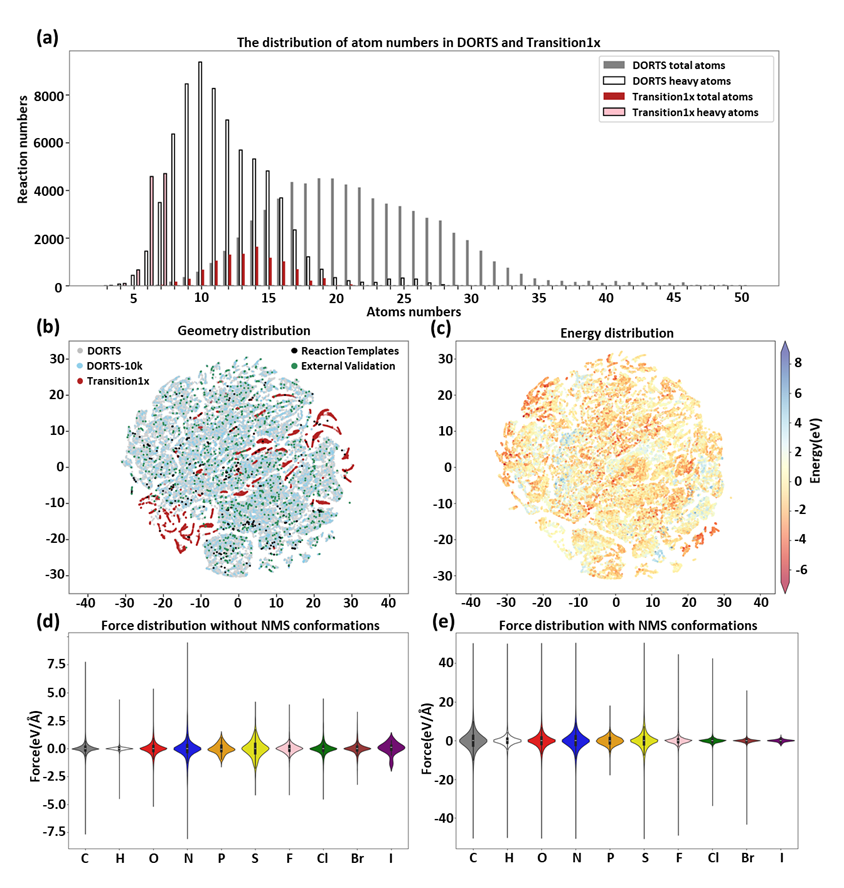
图 2. DORTS数据库
模型精度与效率
DeePEST-OS模型结合了Δ-learning策略与高阶等变消息传递神经网络MACE架构,实现了较高的预测精度与效率,如图3所示。DeePEST-OS的能量MAE为0.266 kcal/mol,力MAE为0.380 kcal/(mol·Å)。在消融实验中,若去掉Δ-learning模块,MAE为1.112 kcal/mol,力MAE为0.819 kcal/(mol·Å),精度大幅下降。若使用其他模型架构(如PaiNN),MAE为2.948 kcal/mol,力MAE为3.485 kcal/(mol·Å),精度大幅下降。在苯酚硝化反应的测试中,DeePEST-OS在单核CPU上的过渡态搜索耗时仅为9.3秒,而DFT方法需要8051秒,速度提高近千倍。此外,DeePEST-OS在保持高效的同时,还能确保三维结构几何精度。关键结构参数分析表明,DeePEST-OS重建的IRC路径中N-O键长与N-C-H键角的演变趋势,与DFT基准的结构偏差显著小于半经验量子化学方法(GFN2-xTB)方法,展现出“既快又好”的独特优势。
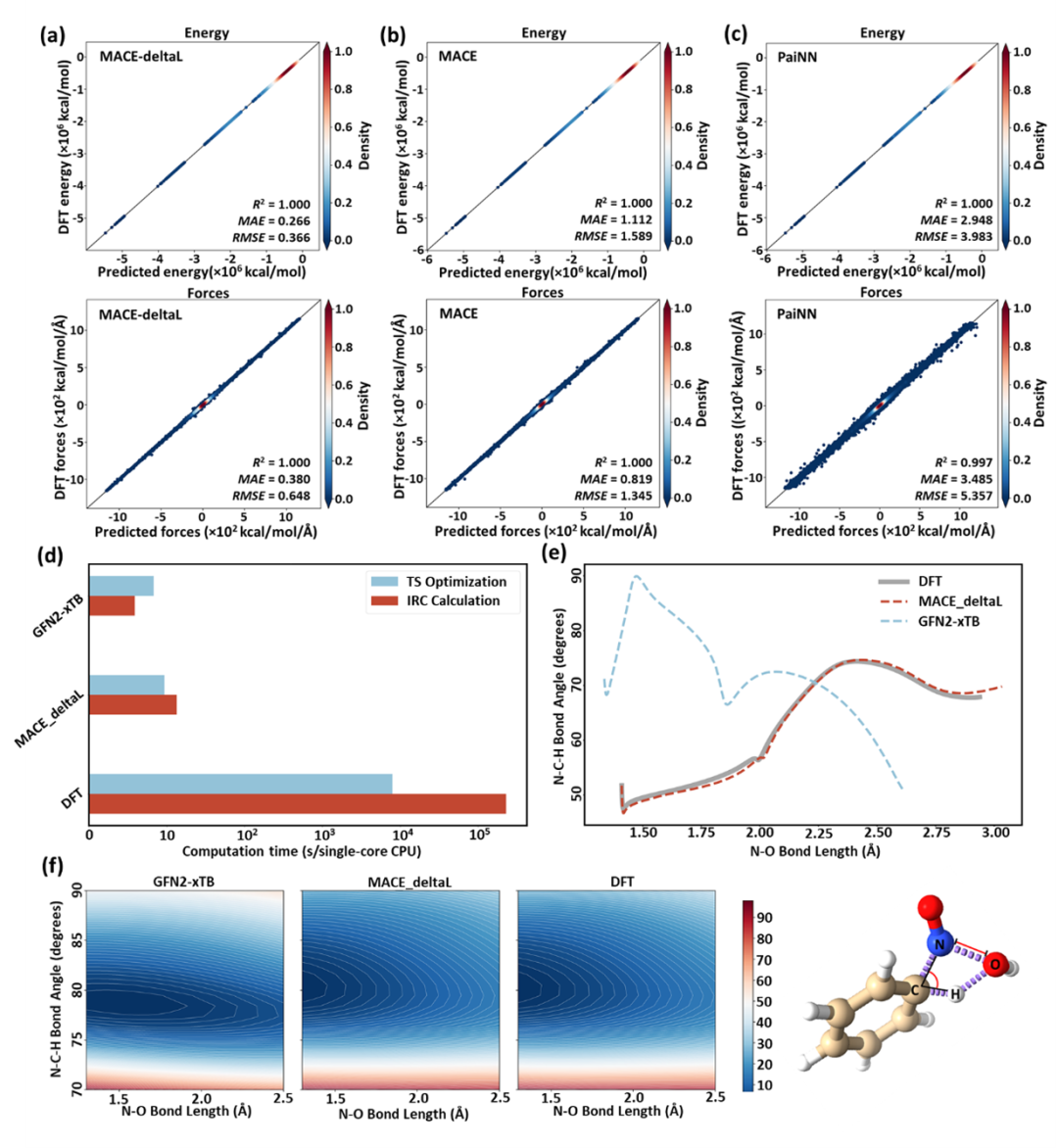
图 3. DeePEST-OS模型预测精度与效率
对比近期同类型模型:React-OT模型
DeePEST-OS模型与近期同类型模型(React-OT)对比如表2所示。
表2 DeePEST-OS对比React-OT
在有机药物分子的研究与开发中,硫、磷以及卤素元素的应用极为广泛。这些元素在增强药物分子的稳定性、生物利用度以及治疗效果等方面具有不可替代的作用。然而,对于现有的React-OT模型(以及2025.6.16发表的DPA-3-DF模型)而言,它们在处理含这些关键元素的化学反应时存在显著局限性,主要原因在于它们使用的反应数据集仅涉及C、H、O、N四种元素。
在计算精度和效率方面,DeePEST-OS模型的优势更加明显。使用相同的数据进行训练和测试(transition1x数据库中9000个反应进行模型训练,其余1073个反应进行外部测试评估),DeePEST-OS在几何结构预测精度和能量计算误差上均优于React-OT模型。此外,DeePEST-OS进行过渡态搜索并计算势垒,在单CPU核上平均每个分子仅需3.4秒即可,而React-OT搜索过渡态则平均每个分子需要V100 GPU 5.8秒,计算势垒则额外平均每个分子需要单核CPU 93.9秒,效率差异一目了然。
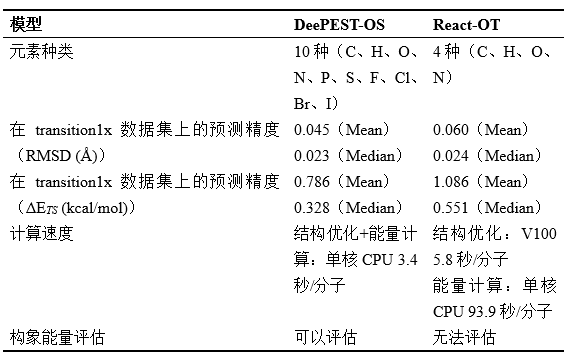
此外,DeePEST-OS能与构象搜索软件(如本团队研发的GENConf软件)联用,精准确定最稳定的过渡态构象异构体,而React-OT模型无法搜索能量最小过渡态构象,如图4所示。通过这种联用,DeePEST-OS可以更高效地探索过渡态构象空间,有助于提高反应动力学模型预测精度。
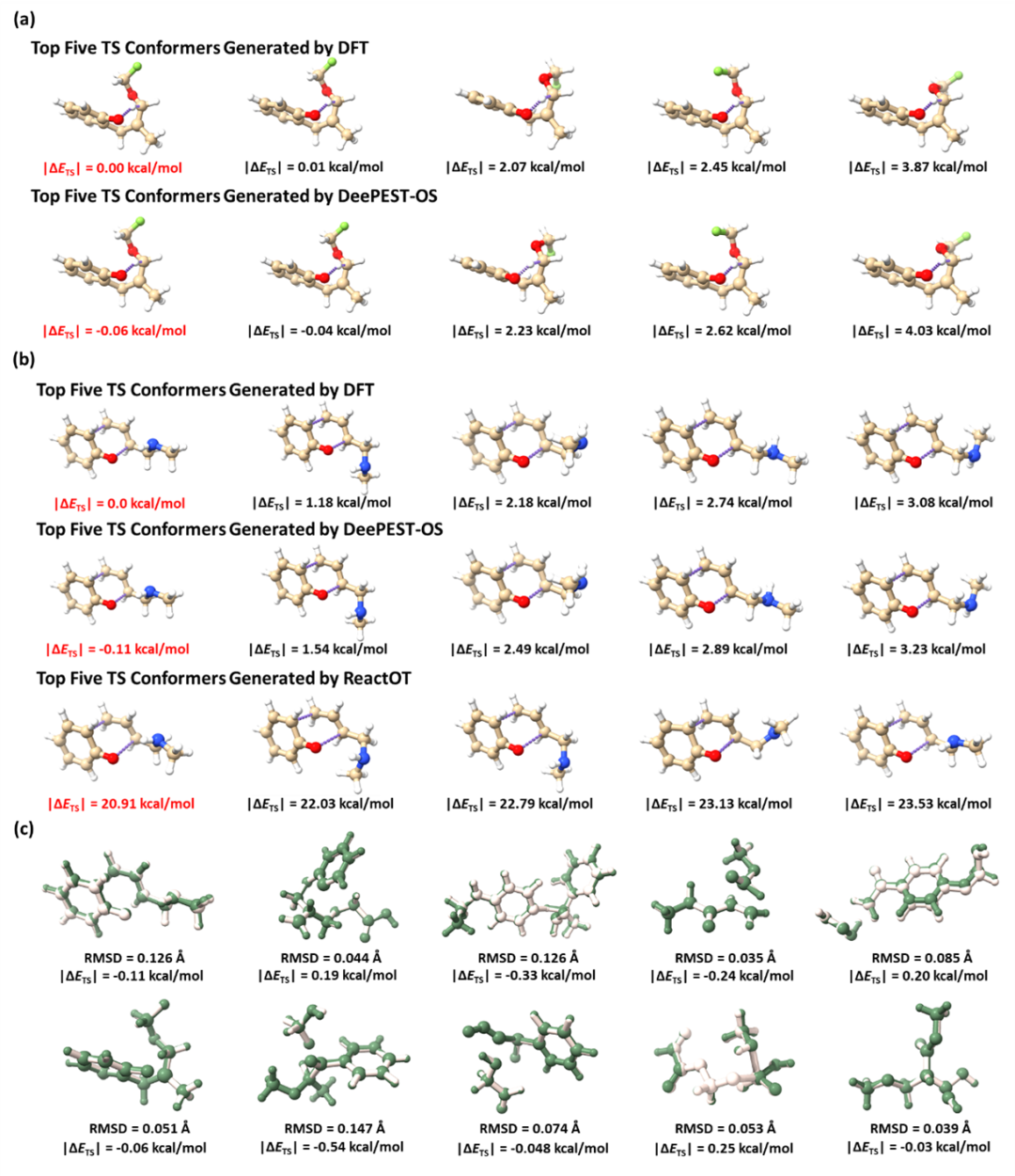
图 4. 能量最小过渡态构象搜索
集成DeePEST-OS模型与药物逆合成规划算法
DeePEST-OS模型可与逆合成规划算法集成,应用于药物逆合成路线的反应势能面预测,从而评估合成路线的反应动力学速率。以扎托司琼(Zatosetron)药物为例,本研究使用课题组自主研发的RetroSynX逆合成规划软件,设计了Zatosetron合成路线,进一步使用DeePEST-OS精准筛选最稳定中间体/过渡态构象并计算基元反应能垒,如图5所示。数据显示,数据显示,DeePEST-OS在完整反应路径中保持平均几何结构偏差(RMSD)0.16 Å、反应能垒绝对误差(ΔETS) 0.43 kcal/mol的卓越精度,相较GFN2-xTB方法实现数量级提升。更重要的是,在预测TS_3rd_2/TS_4th(涉及51个原子)等超越DORTS数据库中原子数最多的分子以及React_3rd_b这种数据库中不存在的几何结构时,模型展现出超越训练数据记忆的化学推理能力,通过原子作用模式自主解析成功预测未见过渡态结构,为突破计算化学数据边界提供了智能解决方案。这一成果不仅彰显了DeePEST-OS在处理复杂有机合成路径方面的强大能力,更凸显了其在反应类型和复杂度上的独特优势。在该案例中,React-OT和DPA-3-DF模型受化学元素种类限制而无法适用。
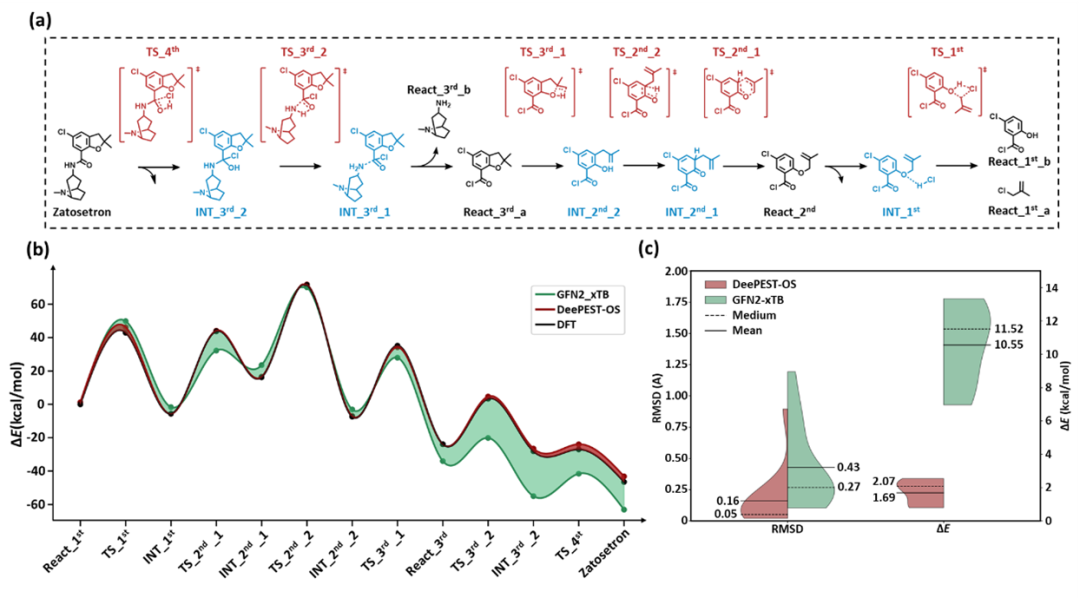
图 5. DeePEST-OS应用于Zatosetron药物逆合成路线反应势能面预测
未来展望:引领化学反应预测进入智能化时代
DeePEST-OS凭借其全面的数据库、卓越的模型精度和高效的计算能力,为有机药物分子的研究与开发提供了强大的支持。它不仅能够处理复杂的化学反应体系,还能显著提升研究效率,为化学研究和工业应用带来前所未有的便利和优势。未来进一步通过融合主动学习与预训练-微调策略,降低数据构建成本,有望建立更强大的“量子精度-高通量筛选”协同新范式,推动合成化学向智能化设计时代大步迈进,为科学研究和工业应用插上智能化的翅膀,开启化学研究的新纪元。
该论文第一作者为大连理工大学化工学院,任凯派博士生和唐坤博士生。
通讯作者为大连理工大学化工学院,刘奇磊副教授。个人主页:https://faculty.dlut.edu.cn/liuqilei/zh_CN/index.htm。
参考资料
Ren K, Tang K, Zhao Y, Zhang L, Du J, Meng Q, et al. DeePEST-OS: A Generic Machine Learning Potential for Accelerating Transition State Search in Organic Synthesis. ChemRxiv. 2025; doi:10.26434/chemrxiv-2025-mzz6w This content is a preprint and has not been peer-reviewed.
内容中包含的图片若涉及版权问题,请及时与我们联系删除

评论
沙发等你来抢