
DRUGAI
批次效应显著干扰了多个单细胞实验之间的比较。现有的批次效应去除和量化方法主要关注于跨批次的细胞对齐,往往忽略了基因层面的批次效应。研究人员提出了一种量化单个基因批次效应的新指标:Group Technical Effects(GTE)。通过GTE分析发现,批次效应在数据集中对不同基因的影响存在不均。某些高度敏感的基因(HBGs)在不同数据集中不尽相同,并在批次效应中起主导作用,而非HBGs则表现出较低的批次效应。研究人员还发现,仅移除三个HBGs就足以显著引入批次效应。此外,GTE方法还可以用于评估细胞层面的批次效应,其性能优于现有量化方法。研究还观察到,生物学上相似的细胞类型在批次效应上也表现出相似性,这为制定更有效的数据整合策略提供了依据。GTE方法具有良好的适用性,能够扩展至多种单细胞组学数据。

随着单细胞测序技术的快速发展,研究人员获得了前所未有的关于细胞状态和组织复杂性的洞察力。然而,多批次实验设计在实际应用中不可避免,如多时点采样、不同实验人员或平台执行的重复实验等,导致数据中不可避免地掺杂技术性变异,即批次效应。这种非生物学的偏差不仅干扰下游分析,还会掩盖真实的生物信号。
尽管目前已有大量方法用于校正批次效应,包括早期针对微阵列数据的模型与近年来发展出的深度学习整合框架,但大多数策略聚焦于将不同批次的细胞嵌入至统一的低维空间,而较少关注特定基因在各批次间的表达一致性问题。已有研究指出,批次效应可能对某些基因或特征产生强烈影响,但在不同特征间的分布与模式仍缺乏系统性的量化和刻画。
为弥补这一空白,研究人员构建了GTE指标,旨在从基因维度出发,精确衡量其受到的批次影响,并探索其在单细胞组学多数据模态下的广泛适用性与整合价值。
结果
GTE模型框架与概念
GTE指标的核心思想源于变异性分解。研究人员假设,如果一个基因在某一细胞类型中不受批次效应干扰,则其在所有细胞中的表达波动应主要来自生物内在变异;相反,如果批次引入了系统性扰动,则该基因的总体变异将被放大。GTE通过比较同一细胞类型内“批次合并变异”与“批次分组变异”来估算技术性噪声的大小,从而量化该基因的批次敏感性。全数据集中的GTE总和即可用于判定全局批次影响的主导基因群体——即HBGs。
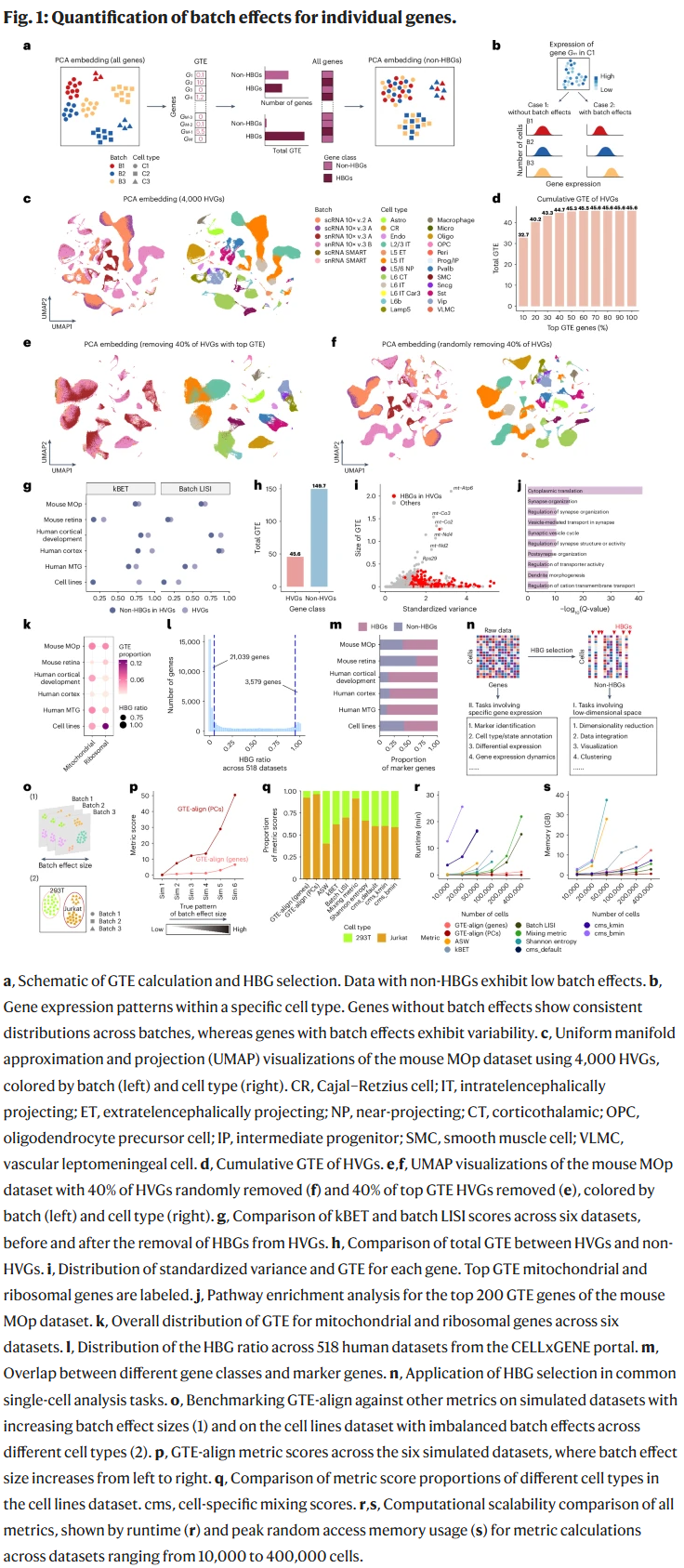
单细胞数据中的批次效应分布模式
在小鼠初级运动皮层数据集中,研究人员首先在4,000个高变异基因(HVGs)上应用GTE计算,发现仅排名前40%的HVGs就贡献了约98%的总GTE。将这些基因剔除后,数据集的主成分分析显示细胞类型分布更清晰,批次间的混合效果显著提升。作为对照,随机移除相同数量的HVGs则未能有效缓解批次效应。
进一步在五个额外单细胞数据集中验证后,研究人员一致观察到:非HBGs构成的特征空间始终表现出较低的批次效应,并能保持良好的生物学区分度。这一结果也得到了其他评价指标如kBET与LISI的支持。
HBG的组成特征与数据集间变异性
研究人员观察到,线粒体基因与核糖体基因在多个数据集中反复出现于高GTE基因集合中。以小鼠MOp数据为例,13个线粒体基因和107个核糖体基因分别贡献了5.9%与5.6%的总GTE,且几乎全部被识别为HBGs。这一现象在其他数据集中也得到一致验证,说明这些结构性基因对技术扰动尤其敏感。
在分析CELLxGENE平台上收录的518个人类数据集时,研究人员进一步识别了3,579个在人群中普遍敏感的HBGs,以及21,039个普遍稳定的非HBGs。尽管GTE值在数据集间高度相关,但HBGs的重合度较低,提示批次效应的作用模式具有明显的数据集特异性。
HBG的选择对下游分析的影响
研究人员指出,仅移除极少量(如3个)HBGs即可引入显著批次效应,反映出这些基因在技术扰动中的放大效应。同时,部分HBGs在功能上与生物标记基因重合,若盲目去除可能丢失关键生物信息。因此,研究人员建议:对于涉及低维空间构建(如降维、聚类、整合)的任务,可优先使用非HBGs特征子集;而对于依赖原始表达模式的任务(如差异分析、标注等),则应保留全部基因表达数据以避免偏差。
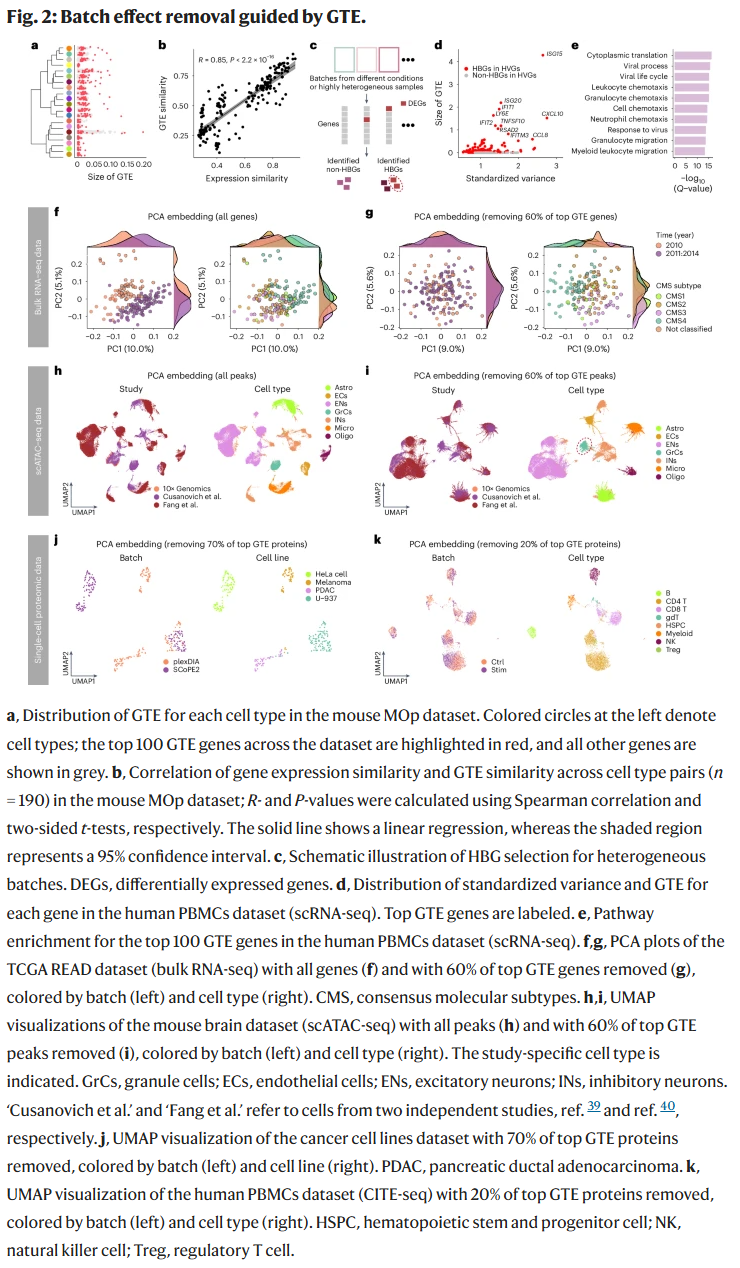
讨论
研究人员首次系统性地提出并量化了单细胞数据中“单基因层面”的批次效应,借助GTE这一框架,不仅揭示了批次效应在基因间的不均性,也为特征筛选与数据整合提供了新的理论基础。研究指出,HBGs与非HBGs在批次响应模式上存在显著区分,且移除HBGs可有效降低批次影响,从而增强细胞类型辨识与空间结构恢复的准确性。
相比传统仅基于细胞嵌入空间的校正方法,GTE关注的是特征层面的结构稳定性,因而可作为前处理步骤,与现有整合方法互补而非替代。对于那些依赖特定基因表达的任务,如细胞类型注释、通路分析或基因动态建模,保留完整表达谱依然至关重要。
此外,GTE所引申出的细胞层面度量——GTE-align,在多个模拟与真实数据中均表现出更高的灵敏性与泛化能力,尤其在不平衡批次设计下表现尤为突出。研究人员还发现,该方法在计算效率上优于多数现有指标,即使在大规模数据(超过40万细胞)中也能实现快速评估。
尽管如此,GTE也存在一定的局限。例如,它高度依赖于准确的分组信息,若细胞注释存在偏误,则HBG识别可能失真。此外,在批次影响广泛弥漫、波及大多数基因的情况下,仅通过去除HBGs仍难以完全消除干扰,需要进一步结合整合技术。特别是在异质性样本或跨条件整合中,若不加区分地移除条件特异基因,可能会影响关键生物亚群的识别,导致潜在生物信号的丢失。
综上所述,GTE不仅为批次效应的量化提供了新路径,也在多组学平台下展示出强大的适应性与理论价值。研究人员认为,该方法未来将在单细胞数据整合标准制定、批次校正工具开发以及跨条件整合任务中发挥重要作用。
整理 | WJM
参考资料
Zhou, Y., Sheng, Q., Wang, G. et al. Quantifying batch effects for individual genes in single-cell data. Nat Comput Sci (2025).
https://doi.org/10.1038/s43588-025-00824-7
内容中包含的图片若涉及版权问题,请及时与我们联系删除

评论
沙发等你来抢