
DRUGAI
深度学习方法在RNA二级结构预测中表现出色,但在未见的、分布外的RNA家族上普遍存在泛化能力不足的问题,这限制了其准确性和稳健性。为此,研究人员构建了一个碱基对基序库,系统枚举了所有本地相邻的三邻碱基对组合,并通过从头建模获取每种基序的热力学能量。在此基础上,研究人员提出了一种深度学习预测方法BPfold,能够学习RNA序列与其对应的碱基对基序能量图之间的关系。在基于序列和基于家族的数据集上,BPfold在准确性和泛化性方面均显著优于其他先进方法。该研究为融合物理先验与深度学习技术在RNA结构与功能发现方面提供了有力工具。

RNA二级结构通过碱基配对决定其三级结构的形成,在催化活性、调控功能以及剪接事件等生物过程中扮演着关键角色。二级结构通常由茎区和环区构成,其中茎区是连续的碱基配对,而环区则由未配对碱基组成。环区中常见多种结构基序,如四环、亲吻环、折角、G-四链体等,这些基序由非标准碱基配对和极性相互作用稳定。
准确获取RNA的二级结构对于理解其与其他生物分子的相互作用具有重要意义,是药物设计和RNA治疗的关键步骤。传统化学探针技术如SHAPE可用于推断二级结构,但高效、纯计算预测方法的快速发展使得基于序列的结构建模成为可能。
早期方法包括同源序列比对与热力学模型,但在缺乏已知家族或能量参数的情境下往往表现不佳。浅层机器学习方法虽然在嵌套结构中有效,但难以预测复杂结构如伪结和非标准碱基配对。相比之下,深度学习方法因其高效性和准确性在近年来受到广泛关注,例如SPOT-RNA、UFold和MXfold2等。然而,这些方法在面对未见家族和数据分布时仍存在显著性能下降,暴露出过拟合和数据不足问题。
由于缺乏高质量、覆盖全面的RNA结构数据集,如何在数据稀缺条件下开发可靠、可泛化的深度模型,成为RNA结构预测领域的关键挑战。
结果
方法概述
研究人员提出BPfold模型,通过引入完整三邻碱基对基序及其热力学能量,有效缓解了数据覆盖不足问题。BPfold的核心在于两部分:
碱基对基序能量:定义了包括邻近碱基的标准碱基对,通过从头建模计算其热力学能量,并建立基序库,用于任意输入序列的能量图生成,从而为深度学习模型提供热力学先验。

碱基对注意力机制:设计了融合Transformer与卷积模块的注意力块,将RNA序列特征与基序能量图联合建模,提高了对碱基对相互作用的表达能力。
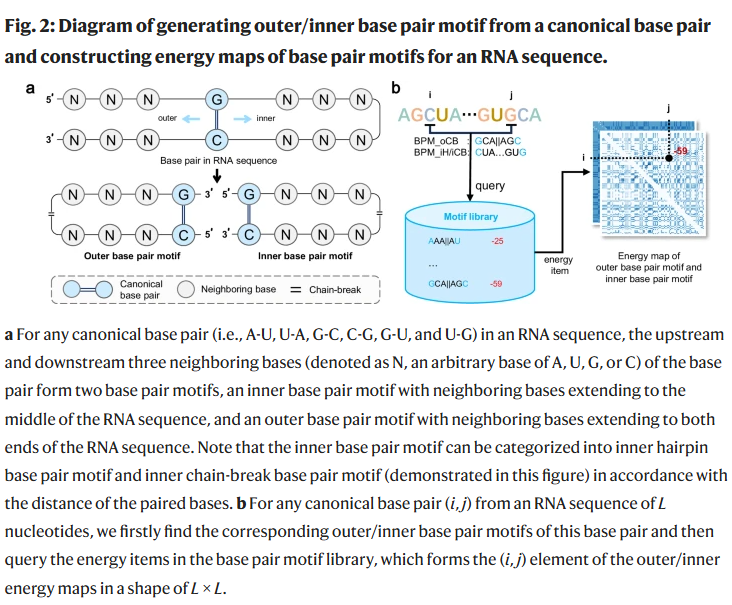
构建基序能量库
研究人员将碱基对基序分为三类:内发夹型、内断链型与外断链型,并采用BRIQ方法从头建模三邻基序的三维结构,结合量子力学与统计能量评分,生成统一归一化的能量分数。该库共包含75,990种标准碱基对组合,涵盖A-U, G-C, G-U等配对。每个输入RNA序列都可被映射为两个L×L维度的能量图(分别对应内外基序),作为神经网络的输入之一。

验证基序能量的有效性
通过消融实验,研究人员分别移除或仅保留不同类别的基序能量,发现完整引入所有基序能量(BPM)时,BPfold在所有指标上显著优于其他配置(如F1值提升至0.689)。尤其在五类RNA家族(如Cobalamin、Twister、RAGATH-18等)上,完整模型的提升尤为明显,表明热力学能量显著增强了模型对未知家族的泛化能力。

序列级评估
在bpRNA-TS0与ArchiveII两个广泛使用的数据集上,BPfold均取得最优成绩。与传统方法(如RNAfold、LinearFold)和其他DL方法(如SPOT-RNA, MXfold2)相比,BPfold在F1、INF等指标上领先。例如在bpRNA-TS0上,BPfold F1值为0.658,显著高于SPOT-RNA(0.625)和MXfold2(0.575)。
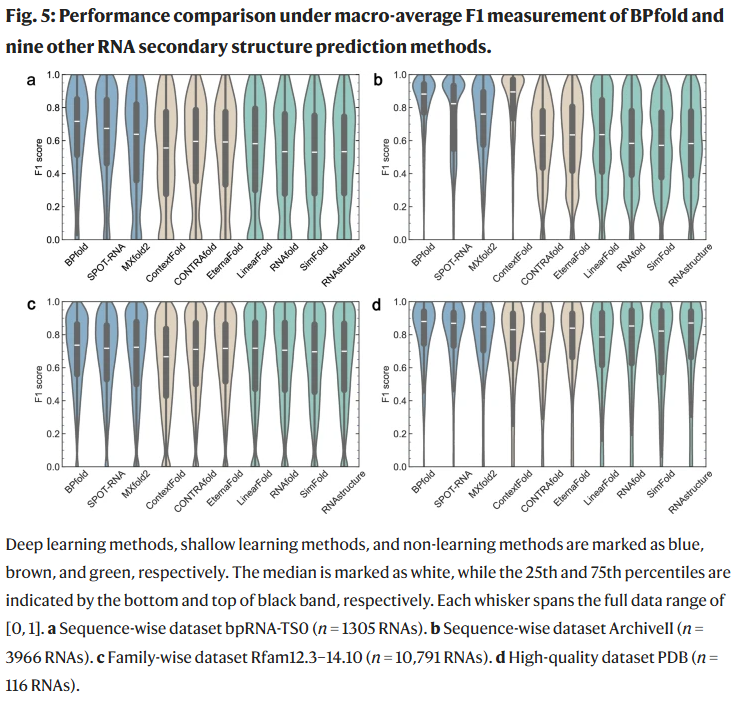
家族级泛化评估
在Rfam12.3–14.10等分布外数据集上,BPfold依然表现最优。即使在非DL方法如EternaFold也具有较高性能的场景下,BPfold仍能凭借热力学基序信息取得最优F1(0.689)与INF(0.694)。在高质量的PDB数据集上,BPfold同样表现出色(F1为0.814),能有效识别密集标准配对。
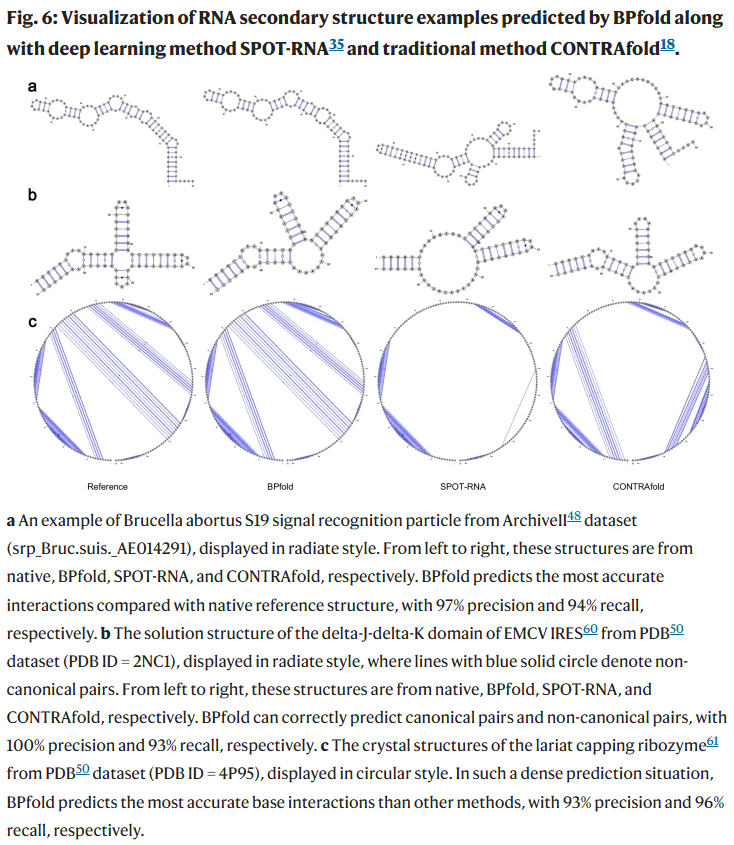
可视化分析与预测速度
BPfold可在10秒内预测不超过1000nt的RNA结构,在1851nt长度下也仅需40秒,速度接近MXfold2和CONTRAfold,远优于SPOT-RNA。此外,可视化结果显示BPfold预测结构与真实结构高度一致,特别是在长序列与非标准配对场景中,展现出良好的建模能力。
构建置信指数
为提升预测结构的可解释性,研究人员设计了基于网络输出前后差异的置信指数,并与F1分数之间具有显著正相关(Pearson系数达0.728),可用于无标签样本的结构可靠性评估。

讨论
研究人员通过完整构建三邻碱基对基序库,引入热力学能量图并结合注意力机制,提出了BPfold这一新型深度学习架构,显著提升了RNA二级结构预测在泛化性、准确性与计算效率上的综合能力。其核心创新点在于:
从结构层级弥补数据不足:以基序为单位替代全结构,显著拓展了有效训练样本空间;
融合物理先验与数据驱动建模:将可解释的能量评分直接注入神经网络输入,提升对真实结构的感知;
具备良好可拓展性与可移植性:碱基对能量图可作为模块集成至其他深度架构如UFold、CONTRAfold等;
面向长序列、非标准配对、少样本等挑战提供思路:如引入动态位置编码、构建置信评估机制。
尽管BPfold在多个指标上领先,但仍存在挑战,包括:扩展至更长邻域的基序建模需较高计算成本;非标准配对数据稀缺,导致预测难度高;长序列数据分布不平衡,限制模型泛化。未来可考虑通过少样本学习、领域适应、半监督等策略进一步突破。
总之,BPfold展示了深度模型融合物理能量先验的新范式,为RNA结构预测及功能解读开辟了新路径。
整理 | WJM
参考资料
Zhu, H., Tang, F., Quan, Q. et al. Deep generalizable prediction of RNA secondary structure via base pair motif energy. Nat Commun 16, 5856 (2025).
https://doi.org/10.1038/s41467-025-60048-1
内容中包含的图片若涉及版权问题,请及时与我们联系删除

评论
沙发等你来抢