
G蛋白偶联受体(G protein-coupled receptors, GPCRs)是药物研发中的重要靶点,约三分之一的FDA批准药物针对该蛋白家族。
2025年7月3日,来自Tri-I TDI、薛定谔公司以及加利福尼亚大学圣迭戈分校的研究人员在npj drug discovery上报告了AI与物理学相结合的计算方法在GPCR药物发现的最新进展,重点探讨了AI驱动的蛋白质结构预测如何在各个阶段推动GPCR药物发现。
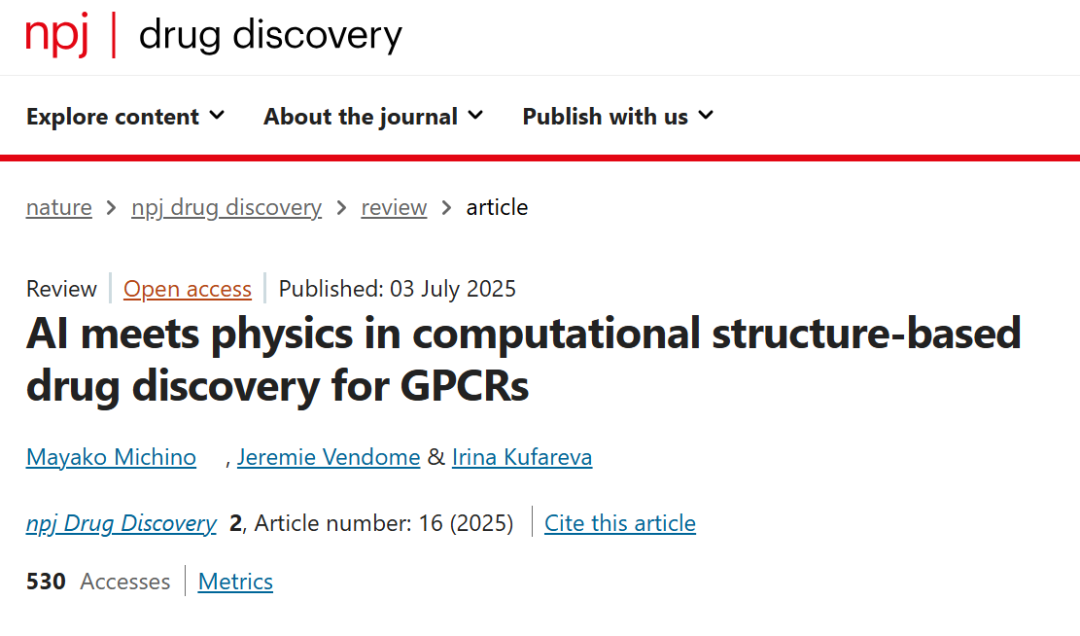
SBDD流程概述
基于结构的药物发现(Structure-based drug discovery, SBDD)利用蛋白靶标的三维结构确定及优化临床阶段前的化合物。
SBDD的四个关键环节如下:1)受体建模,构建或选择靶标受体的三维结构模型;2)配体结合受体复合物建模,生成配体构象及受体适合配体结合的构象;3)hit筛选,发现初始活性化合物;4)hit到先导化合物的优化,优化化合物的效力和药物性质。
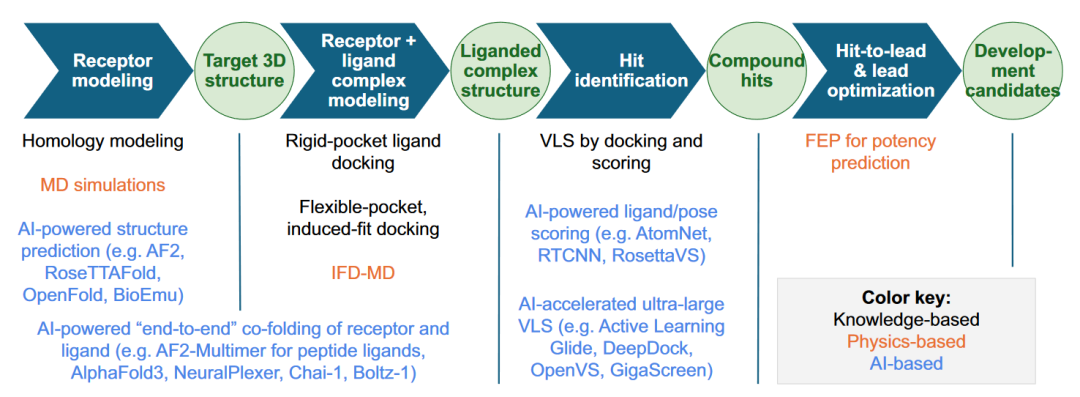
图1 基于结构的药物发现关键环节及方法
GPCR结构预测
靶标蛋白在相关功能状态下的精确三维结构是SBDD的核心组成部分。然而,对于GPCRs来说,高分辨率的实验结构稀缺,限制了SBDD的应用。
自2020年以来,人工智能方法在蛋白质结构预测方面取得了突破性的进展,如AlphaFold2(AF2)和RoseTTAFold在CASP14竞赛中的表现接近实验精度。这些基于人工智能的结构预测算法都是在蛋白质数据库(Protein Data Bank, PDB)中已知的实验结构上训练的,因此如果没有PDB中实验GPCR结构数量的爆炸性增长,GPCR就不会成功。截至2025年3月,实验确定的结构已经解决了大约四分之一的GPCR超家族(约800个GPCRs中有235个),但AF2模型可用于所有超家族成员,包括其最大的子家族Class A(674受体, 图2)。
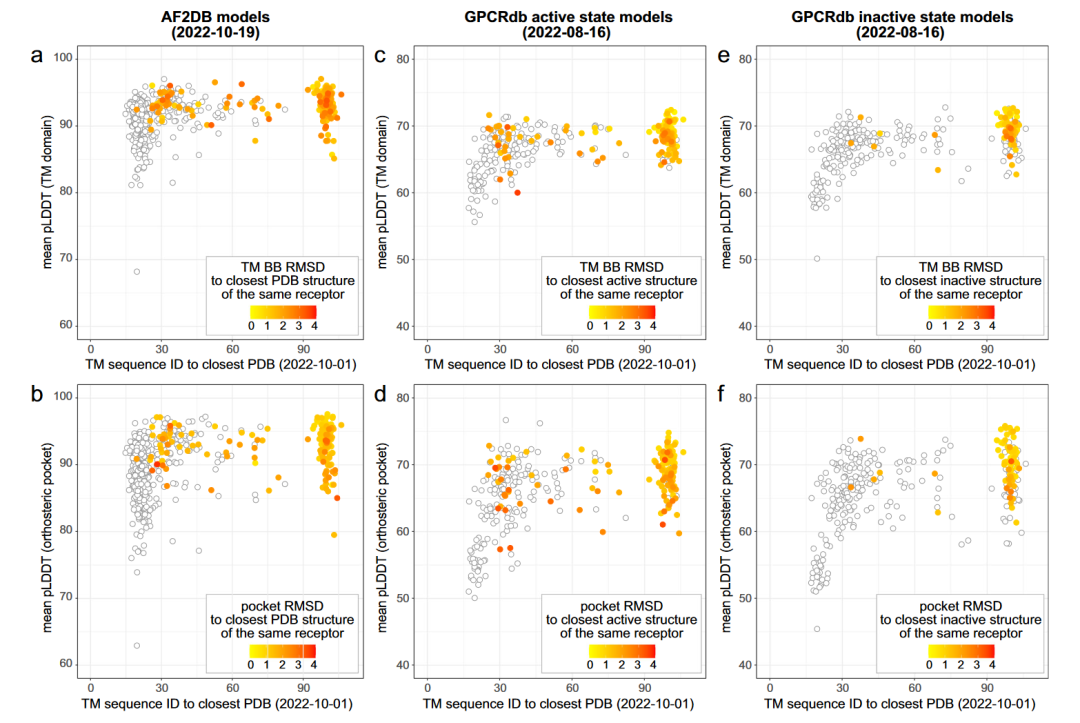
图2 Class A GPCR AF2模型的置信度以及几何精确度
一些研究系统地检验了AF2和RoseTTAFold预测的GPCR模型的几何精度。He等人的研究表明AF2在预测GPCR结构时,虽然跨膜域整体准确,但在胞外环与跨膜域的连接、信号转导蛋白结合区域以及结合位点氨基酸侧链的精确构象上存在不足,影响了配体结合和功能状态的准确模拟。Lee等将AF2和RoseTTAFold对73个GPCRs的预测模型与实验结构进行了比较,发现AF2比RoseTTAFold更精确,而对于没有良好模板的受体,两者都比传统的同源模建方法表现更好。这些发现与专门从事GPCRs实验结构测定的实验室的经验一致,也与对于AF2在不同蛋白家族的预测结果的总体评价一致。此外,尽管AF2模型具有"接近实验精度"的声誉,但Terwilliger等人的结论是,在确定的结构中,预测模型的平均误差高于实验误差。除了几何精度外,另一个重要方面是它们的物理有效性,这涉及到共价键(如键长、角度和芳香环形状)和非键分子内相互作用(如空间冲突)。在AF2的情况下,非物理接触和几何形状通常存在于初始模型中,可以通过预测过程中的模型松弛来消除。
AF2的一个主要局限在于它无法直接建模靶标蛋白功能性不同的构象状态。例如,GPCR在与激动剂结合后会发生显著的构象变化,因而至少可以采取两种不同的状态,即非活化态(inactive)和活化态(active)。然而,AF2所预测的模型通常只代表其中一种状态,这种偏向源自其训练数据库中已有实验结构的分布。通过分析TM6和TM7的预测构象(这两个跨膜螺旋是受体活化状态的指标),He等人发现,AF2倾向于对A类GPCR预测平均构象,而对B1类GPCR则更倾向预测活化态构象,这与PDB中这些类的结构状态分布相一致(A类中55%为非活化态,37%为活化态;B1类中70%为活化态)。AF2模型中所呈现的构象变异与局部不确定性,往往与蛋白本身的内在动力学变化是一致的。因此,对于某些GPCR,如果其在PDB训练集中有足够多构象多样的模板,AF2就有可能预测出覆盖部分受体构象谱的构象集合。
为了让AF2可重复地生成特定激活状态的GPCR模型,Feig团队开发了一个扩展方法,称为AlphaFold-MultiState,该方法利用带有激活状态标注的GPCR模板数据库进行建模。生成的模型与此前已知以及后来解析出的处于相应状态的GPCR实验结构高度一致(图2c-f)。此外,也有研究团队通过修改和减少输入的多序列比对深度,成功生成了具有功能相关构象状态的蛋白结构集合。
自AF2发布以来,计算结构生物学界已开发出多个改进实现版本,旨在提升AF2的可扩展性、可访问性和应用性。例如,由AlQuraishi团队主导的OpenFold是对AF2的一种GPU内存优化再实现,可用于在新数据集上进行再训练。MassiveFold则通过并行化大幅降低了AF2的计算时间。最后,为了解决单一构象预测的局限,微软研究院近期推出了BioEmu,这是一种可扩展的生成式深度学习模型,用于生成蛋白的平衡构象集合。不过,以上这些研究方法目前尚未专门应用于GPCR结构预测。
GPCR-配体复合物几何预测
如果模型中配体在结合口袋中的构象接近天然构象,并形成与实验观察类似的受体-配体相互作用,那么它就能在如下方面发挥关键作用:解释和预测构效关系、优化化合物效能、以及发现具有不同骨架的新配体。在基准测试中,通常通过将受体结合口袋或跨膜结构域最佳叠合后,计算配体重原子的均方根偏差,来评估预测配体构象相对于实验结构的准确性(图3a-c)。而受体-配体之间的相互作用,则可通过对比预测与实验结构中所有原子对之间的距离来评估。对于任意一组受体-配体复合物模型,配体的RMSD与正确预测的相互作用比例通常只有较弱的一致性(图3d,e)。因此,通常将这两个指标结合起来,并放在PDB中已知同样复合物的高分辨率结构对比中观察其变化范围,以此评估预测模型的合理性。
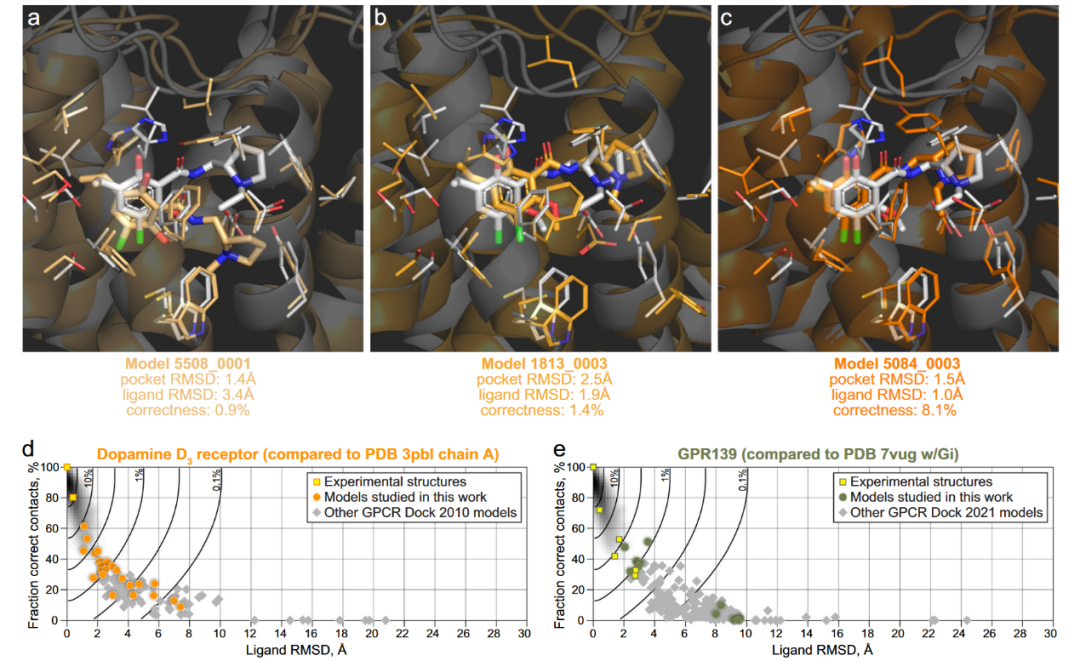
图3 预测GPCR配体复合物模型的几何精度的变化
早期配体对接方法通过将配体柔性构象对接到刚性受体结合口袋,评分并排序配体构象。该对接方法的成功高度依赖于结合口袋的准确性,以及其形状是否适配于所研究配体。它还取决于配体的类型,例如具有多个可旋转键的配体(如肽类)更具挑战性。在对接中固定受体结构有利于计算效率,但会忽略受体在配体结合时可能发生的重要构象重排,即诱导契合。随后,研究人员发现,AF2虽然提高了GPCR结构预测精度,但仍然难以直接用于高精度配体对接。经过一系列尝试(如对结构进一步优化、加入功能状态信息、引入侧链柔性等),最终AF2模型的表现能优于传统同源建模。
GPCR Dock是权威的结构预测比赛,检验AF2等工具在真实盲测中的表现。GPCR Dock 2021发现,AlphaFold-Multimer所实现的共折叠策略在预测大分子天然肽类配体复合物的几何结构方面,基本解决了包括受体诱导契合和配体柔性等关键挑战。然而,对于小分子配体复合物,这些问题仍未得到解决。因此,计算结构生物学界从那以后一直在探索类似的方法能够准确预测小分子配体在受体中的构象,以及受体口袋结构的相应重排。
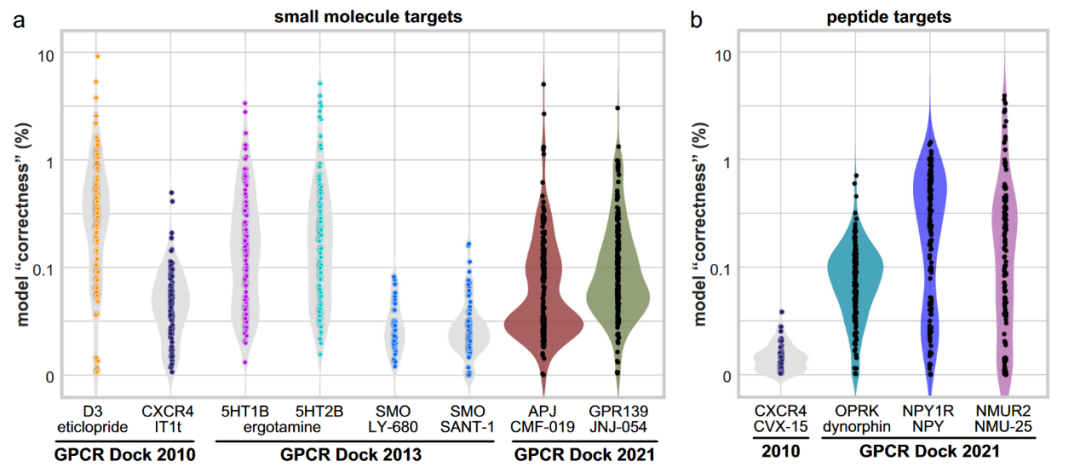
图4 GPCR Dock竞赛中预测GPCR-配体复合物模型几何准确度分布
近几年间,多个用于与小分子化合物和非蛋白质生物分子(如离子、核酸)形成复合物的端到端方法陆续发布,代表方法包括DiffusionProteinLigand,NeuralPlexer, RosettaFold-AllAtom,以及AlphaFold3(AF3)。这些新方法均基于扩散模型的共折叠,其目标是克服之前由于受体结合位点不准确或构象不合理,导致配体对接效果不佳的问题。考虑到AF3的使用条款限制,许多学术机构和产业团队纷纷开发了自家版本,包括ChaiDiscovery的Chai-1,Iambic Therapeutics的NeuralPlexer 2和3 beta,Ligo Biosciences的AlphaFold3,百度的HelixFold3,字节跳动的Protenix,Umol,Boltz-1。
不同共折叠方法在预测受体-配体复合物几何结构方面的准确性差异很大,并且高度依赖所使用的评估基准。另一个关键问题是预测结构的物理合理性。基于扩散模型的方法经常生成一些违反键长和键角限制的非物理模型,还可能出现分子内或分子间的立体冲突,这很可能是由于训练过程中对特定数据子集的过拟合所致。与AF2不同,这些非物理错误并不能总是通过模型松弛有效去除。与这些观察一致,作者尝试使用扩散共折叠方法复现部分GPCR Dock复合物结构时,也取得了好坏参半的结果,部分模型中存在非物理性的受体内部或受体-配体相互作用(图5a,c),并且预测的几何精度也存在较大波动(图5b,d,e-j)。
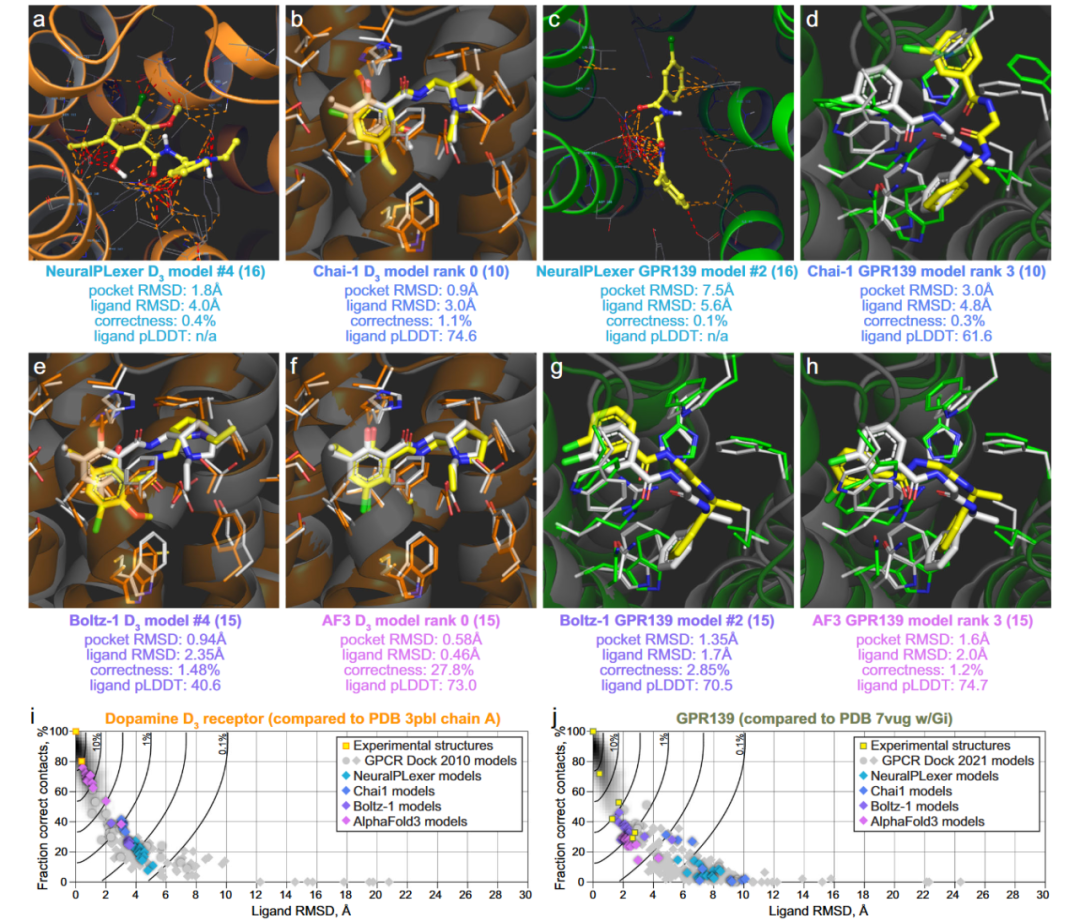
图5 dopamine D3 receptor-eticlopride与GPR139-JNJ复合物的模型预测结果
考虑到当前AI端到端复合物结构预测的局限,基于物理的建模方法依然是预测GPCR与小分子配体复合物结构(特别是建模诱导契合)的关键工具。此类方法包括:集成对接与打分以及IFD-MD方法论(结合蛋白结构预测、基于药效团的配体对接、刚性受体对接及显式溶剂分子动力学模拟)。研究还报道了将传统力场打分方法与深度学习结合,进行配体构象预测与优化的成功案例,这表明人工智能与物理建模的协同或许是未来的最优路径。
配体发现
在药物发现流程中的初筛阶段,目标是找到具有新颖骨架结构,并对靶标蛋白表现出可测效力和所需药效的化合物。计算方法主要包括两类:基于配体的方法和基于结构的虚拟配体筛选。基于配体的方法利用已知化合物的二维或三维药效团进行筛选,这种方法往往只能发现与已有结构相似的新化合物,因此在骨架多样性和化学类型上存在局限。而基于结构的虚拟配体筛选(structure-based virtual ligand screening,VLS)依赖一个或多个靶点受体结合口袋的三维结构模型,能够发现更多结构新颖、类型多样的配体。VLS的成功率在很大程度上取决于所使用的三维模型的质量。对于GPCRs,如果使用高分辨率实验结构进行VLS,hit率可高达约60%。而基于计算预测结构模型的VLS,一般来说hit率较低,筛出的配体亲和力也较弱。
回顾性虚拟筛选(retrospective VLS)是目前评估结构模型在VLS中表现的主流方法。具体而言,让模型区分少量已知活性化合物和大量诱饵化合物,其中诱饵化合物与活性分子在理化性质(如大小、电荷、柔性)上相似,但在结构上无关。常用的构建诱饵集合的工具是DUDe数据库。通过比较模型在该任务中对已知活性物质与诱饵的打分能力,便可评价模型的虚拟筛选表现。这种能力不仅可以用来预测模型在前瞻性筛选中的成功率,而且据报道它还与模型能否预测出活性分子的几何正确构象相关。但模型可能对已有骨架有偏向,不利于新骨架的发现。因此,实际筛选中建议引入一些未经回顾性验证的新模型以提升化学多样性。
考虑到AF2所预测的GPCR-配体复合物在几何结构上的准确性已有显著提升,人们对其在基于结构的先导化合物筛选中取得更好成功率也抱有期望。已有研究结果表明,未经优化的AF2模型倾向表示apo状态,结合位点结构往往是坍塌的,因此并不适合直接用于VLS。通过侧链重建、结合位点建模等方法对AF2模型进行精修后,能显著提升筛选效果。另外,由Lyu等人开展的大规模VLS比较实验发现AF2模型在回顾性VLS中表现较差,且结合位点的侧链构象变化明显,但在前瞻性识别新配体方面依然非常有效。
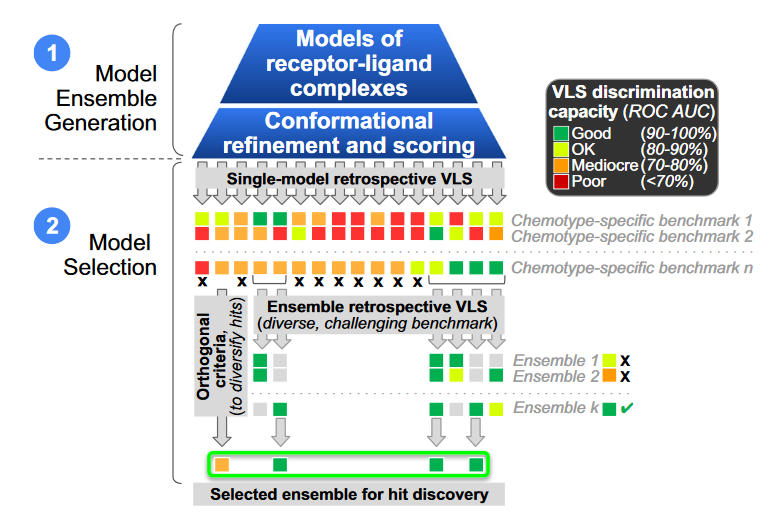
图6 用于前瞻性基于结构的初始化合物发现的模型集生成的最佳实践
打分函数决定VLS排名质量,是虚拟筛选流程中关键但常被忽略的环节。传统的基于物理力场的打分函数,考虑了众多物理因素,如形状互补性(范德华力),电荷互补性,氢键作用,构象应变等。这些基于力场的打分函数在使用高分辨率的实验结构时有优异的表现但对结构误差敏感。随着人工智能技术的兴起,近年来出现了许多基于深度学习的打分函数,包括EquiScore,AtomNet,RTMScore,RTCNN,IGModel, RosettaVS。这类AI打分函数在各种基准测试中,几乎总是表现出优于传统物理模型的筛选效果,主要是因为它们对构象偏差更具容错性,但常存在过拟合与非物理惩罚机制不足问题。这些AI打分方法在GPCR药物发现中的应用尚处初期,但有潜力提高对建模误差的鲁棒性。
除结构预测与复合物打分外,AI也越来越多地被用于满足对超大规模化合物库(十亿级)的筛选需求,例如MolPAL,Active Learnig Glide,DeepDock,OpenVS, GigaScreen,CP framework。其核心思路是先对化合物库中小部分进行对接与打分,利用结果训练一个靶点特异的神经网络,基于化合物2D结构预测结合分数,随后将神经网络应用于整个数据库,再对打分靠前的小部分化合物进行重新对接与打分。重复此流程直至收敛或神经网络出现过拟合迹象。这种AI加速流程在膜蛋白筛选中已被证明其强大潜力。不过,目前尚未见到它在GPCR上的前瞻性应用实例。
近年来,越来越多的研究开始使用生成式AI进行配体发现,这种方法可以绕过传统化合物库筛选的挑战,并在先导化合物发现中作为VLS的补充。Powers等人利用SE(3)等变图神经网络,直接在受体结合口袋中生成分子结构。实验结果表明它在多个靶点(包括GPCR)中表现出比传统虚拟筛选更好的配体打分、更好的类药性以及更优的药代动力学性能。另外,Thomas等人以多巴胺D2受体为例发现,生成的分子不仅具有更好的预测结合亲和力,而且在理化性质上与已知D2激动剂呈现明显差异。生成式AI正成为配体发现的新兴工具,它可以直接设计具有理想性质的分子,有望克服传统筛选的瓶颈。
hit到先导及先导化合物优化
在药物发现的hit转先导化合物和先导化合物优化阶段,目标是通过对hit或先导化合物的骨架进行结构修饰,优化药物性质,如靶点结合活性,脱靶选择性,以及药代动力学性质。这一过程预测候选分子的活性或结合亲和力,依赖于构建配体-靶点复合物结构模型进行评估。这类模型必须能识别细微结构变化带来的活性变化。即使是现有的最优AI打分模型也很难对同系化合物准确排序其亲和力。
自由能微扰(FEP)是一种基于分子动力学和炼金变换的物理计算方法,常用于基于结构的结合自由能预测,尤其适用于对配体骨架做微小改动的结构优化。这一过程通过模拟两个配体之间的炼金变换来实现,在溶液中和靶标结合口袋中分别进行,并沿着整个变换路径对复合物构象进行分子动力学取样。目前已实现该方法的软件有GROMACS,YANK,Schrödinger的FEP+。在多类靶点中,使用晶体结构对FEP+进行回顾性验证发现,其在相对结合自由能预测方面的准确性几乎可与实验数据相媲美,但在前瞻性应用中,即用于预测尚未测试的新化合物,FEP的实际预测误差往往较高。
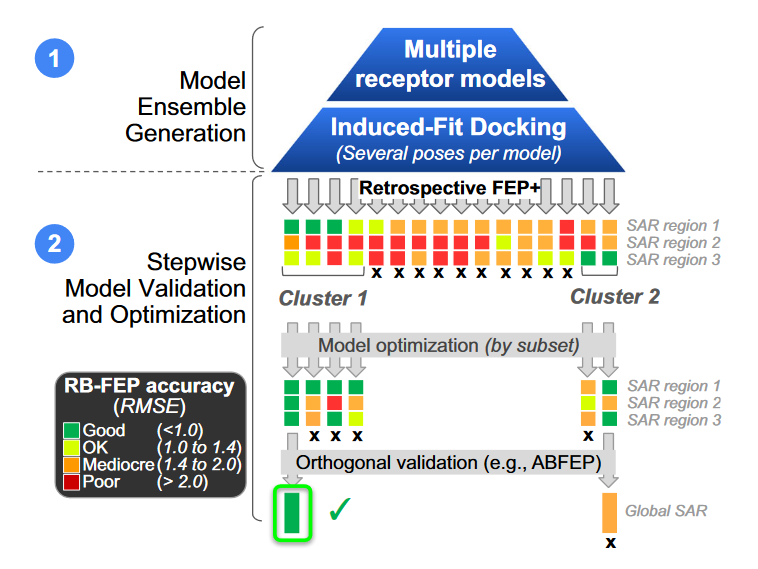
图7 用于先导化合物优化的FEP支持模型的生成与验证最佳实践
近年来,多个研究团队尝试利用机器学习方法来预测相对结合自由能,但预测精度偏低,且泛化能力较差。一些融合了物理知识的AI模型(如PBCNet、IGModel)可能有更好的预测准确性与可解释性。但这些模型的预测精度依然明显低于物理力场方法,如FEP+,可能因为它们忽略了配体结合时结合口袋构象的变化。因此,在亲和力预测领域,基于物理的方法仍是标准。
作者强调使用不充分或不合适的活性数据集进行回顾性RB-FEP评估,可能会误导模型的有效性判断。比如,如果使用的活性数据分布稀疏、范围狭窄,甚至一个几何结构偏差较大的模型也可能得到较高的RB-FEP预测准确性,但其前瞻性应用可能失败。除了RB-FEP,还可以使用绝对结合自由能计算(AB-FEP)来评估模型是否适合用于配体优化的前瞻性研究。对比于RB-FEP只采样配体之间的微小结构变更,AB-FEP则对单个配体在整个口袋中的“出现”和“消失”进行模拟(在口袋和溶液中),另外AB-FEP不依赖同源化合物以及活性数据,只需一个配体即可进行预测。在多个模型都表现出相似的RB-FEP预测性能时,可以通过AB-FEP来进一步优先选择更可靠的模型用于前瞻性研究。
总结
人工智能为用于GPCR的基于结构的药物发现开辟了新途径,极大地提高了高质量结构模型的可获取性。基于AI的共折叠方法也有望解决长期困扰结构建模的诱导契合问题,并为受体与小分子或肽类配体复合物提供准确的结构模型,这对多数药物研发任务至关重要。
然而,这些AI模型在几何精度和物理合理性上仍存在较大差异。另一方面,物理驱动的计算化学方法也在持续进步,包括新的诱导契合对接方法以及高精度的自由能计算(如FEP),它们在GPCR应用中已取得显著成功,而AI所生成的结构模型也进一步拓展了这些物理方法的适用范围。重要的是,几何精度与模型在药物发现中的预测表现并不总是呈正相关。因此,应通过回顾性评估结合恰当的基准体系,来筛选适用于先导化合物发现与优化任务的预测模型。
除了结构预测之外,评分函数的开发及其在虚拟筛选中的加速策略也正在成为AI与物理方法交叉融合的热点,预示着在GPCR药物发现领域更广泛的未来应用前景。
参考链接
https://doi.org/10.1038/s44386-025-00019-0
--------- End ---------
内容中包含的图片若涉及版权问题,请及时与我们联系删除

评论
沙发等你来抢