
蛋白质是组成人体一切细胞、组织的重要成分,其通过打开、关闭、扭曲和重排等运动方式实现生物活性,与其他分子结合或执行复杂功能。传统分析方法如低温电子显微镜、分子动力学(MD)模拟等,虽然能提供高精度的结构信息,但耗时较长且成本高昂。近年来,AlphaFold等模型在蛋白质结构预测方面取得了进展,但仍难以捕捉蛋白质在功能过程中所经历的动态变化。
近日,微软研究院AI for Science团队开发了一种生成式深度学习模型BioEmu,能够以前所未有的效率和精度模拟蛋白质的构象变化。BioEmu能够在一个GPU小时内生成数千种构象状态,其速度显著优于传统MD模拟。此外,通过结合大量的蛋白质结构数据、超过200毫秒的MD模拟数据以及实验测量的蛋白质稳定性数据,BioEmu能以约1 kcal/mol的相对自由能误差准确预测蛋白质的平衡态构象。综上,该模型为理解蛋白质功能机制和加速药物发现打开了新途径。
蛋白质常常会根据相互作用或所处环境在不同构象之间转换,因此解析蛋白质功能一直是一项挑战。微软研究院AI for Science团队的跨学科研究员Frank Noé表示,谷歌DeepMind公司开发的、曾获诺贝尔奖的 AlphaFold 等工具开启了 “结构革命”,为科学家带来了约2亿个静态预测结构,但对于揭示蛋白质动态运动仍显不足。为填补这一技术空白,Frank Noé带领科研团队开发了BioEmu模型。
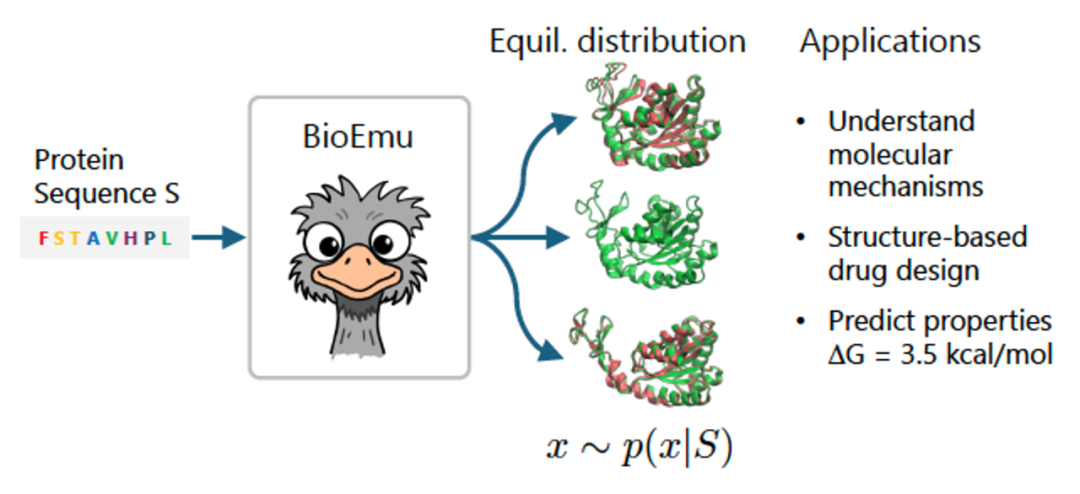
BioEmu基于深度学习神经网络,使用了AlphaFold数据库中的静态结构、超200毫秒的MD模拟数据以及50万条蛋白质稳定性实验数据,并通过多阶段聚类策略进行训练,以学习捕捉真实的平衡行为。利用这些数据,BioEmu生成能够反映蛋白质功能的多样化构象,无需为每个案例运行新的模拟。
特别地,研究团队还为BioEmu设计了一种特性预测微调(PFFT)算法,该算法能使模型的输出结果与实验数据相匹配,即便在缺乏结构数据的情况下也是如此。
Noé解释道:“上述训练使BioEmu能够“预测蛋白质如何移动以及它们在平衡时可以呈现的结构。这是第一个对蛋白质可能具有的不同状态的相对概率进行定量预测的工具。”
BioEmu模型的核心在于将蛋白质序列信息转化为多种可能的三维结构,其通过以下几个关键模块突破传统技术局限:
1.首先采用基于预训练的AlphaFold2模型的蛋白质序列编码器,通过多序列比对(MSA)将蛋白质序列转化为表征信息,为后续构象生成奠定基础;
2.使用粗粒化蛋白质结构表示模块,仅保留蛋白质主链的重原子(即Cα、C、N和O),并将Cα原子Gram-Schmidt正交化处理创造独特的局部坐标系,从而生成蛋白质的主链框架表示;
3.扩散条件生成模型,BioEmu通过逐步去除噪声生成与目标分布相似的蛋白质构象,这种多样化的构象生成方式对于捕捉蛋白质动态行为至关重要;
4.最后是Score模型,其利用多种信息预测分数,从而确保了模型的准确性与稳定性。
此外,BioEmu不能模拟MD,蛋白质与膜、配体的相互作用或温度、
目前,研究团队已在GitHub和HuggingFace上开源了BioEmu模型的参数和代码。
麻省理工学院的博士后计算生物学家Zhidian Zhang表示,过去几年已有多个团队尝试预测蛋白质的替代构象状态,但 BioEmu 的独特之处在于,它能够预测 “不同构象的分布,这是一个难度大得多的问题”。
佛罗里达大学的计算化学家Alberto Perez表示计划在工作中使用BioEmu,并对其开源发布感到欣喜。“当初 AlphaFold 3 未公布代码时引发了诸多不满,因此看到这款工具的代码能够公开获取,我非常高兴。”
参考资料
Sarah Lewis et al. ,Scalable emulation of protein equilibrium ensembles with generative deep learning.Science0,eadv9817DOI:10.1126/science.adv9817
https://cen.acs.org/biological-chemistry/proteomics/Microsoft-AI-predicts-protein-conformations/103/web/2025/07
https://phys.org/news/2025-07-ai-tool-protein-dynamics-aiding.html
内容中包含的图片若涉及版权问题,请及时与我们联系删除

评论
沙发等你来抢