
DRUGAI
今天给大家介绍的是浙江大学药学院康玉副教授和侯廷军教授团队于2025年8月4日发表在Nature Machine Intelligence的一篇论文,标题为“Protein–peptide docking with a rational and accurate diffusion generative model”,第一作者为博士生赵慧锋。
在这项研究中,团队提出了一种基于扩散生成模型的蛋白–肽对接新工具:RAPiDock(Rational, Accurate and rapid Protein-peptide interaction Docking)。该模型通过在平移、旋转、主链与侧链扭转角四个自由度上引入扩散过程,并结合物理约束对肽构象空间进行压缩,有效提升了对接结构物理的合理性与采样效率。同时,RAPiDock创新性地采用了原子-残基双尺度图结构(bi-scale graph),实现对肽分子在原子层级和残基层级的多维建模,进一步增强了模型对结合过程中几何与物理规律的捕捉能力。与现有方法相比,RAPiDock不仅在蛋白结合肽的结构预测任务上实现了准确性和效率的双重突破,还显著提升了对复杂结合构象的识别与建模能力,整体性能超越目前主流的深度学习方法,包括AlphaFold3和AlphaFold2-Multimer。
实验结果表明,RAPiDock在RefPepDB-RecentSet测试集上达到了93.7%的Top-25预测成功率,较AlphaFold2-Multimer提升13.4%,且预测速度提升约270倍(每个复合物仅耗时约0.35秒)。RAPiDock支持对包括多种翻译后修饰在内的92种氨基酸残基进行全原子建模,且具备识别多位点结合构象、还原结合位点关键相互作用与细微构象变化等能力,显著增强了对复杂蛋白-肽体系的建模水平。RAPiDock能快速且精准地生成高质量结合构象,使得基于结构的肽类分子高通量虚拟筛选成为可能。该方法有望成为推动蛋白-肽互作精确解析与高效肽药设计的重要技术支撑。

研究背景
蛋白-肽相互作用(Protein–peptide interactions,PpIs)作为多种生理过程的重要调控机制,具有不可替代的功能。相较于蛋白质或小分子药物,肽类分子因其免疫原性低、合成工艺简单、靶向性强等优势,近年来在靶向治疗、药物递送、分子成像、蛋白功能调控及分子探针等多个生物医药领域得到广泛关注与应用。
实现肽类分子的药物开发与理性设计,首要前提是深入理解其与靶标蛋白之间的结合模式与分子识别机制。然而,蛋白–肽结合通常呈现出弱亲和力、瞬时性强、对环境高度敏感等特性,特别是长度在2~20个氨基酸之间的短肽,其构象相对灵活且易于变化,使得其结构解析面临巨大挑战。结构数据库(RCSB PDB)中的统计数据显示:在已收录的逾22万个蛋白质结构中,蛋白–肽复合物仅占约5.1%,远低于蛋白–蛋白(49.4%)和蛋白–小分子(19.6%)复合物的占比。这一显著差异揭示了实验技术在解析PpI复合物方面的挑战,也反映出该领域在结构数据层面尚存在广阔的研究空白。
在计算方面,主流对接方法主要针对蛋白–小分子体系开发,当应用于PpI体系时,常常面临构象空间庞大、打分函数泛化能力差等突出挑战。以AlphaFold3、AlphaFold2-Multimer(AF2Multi)为代表的深度学习结构预测工具已被尝试迁移用于蛋白-肽复合物预测。但其核心依赖多序列比对(MSA)进行建模,计算成本仍然相对较高,对目标序列的丰富性也有较高要求。更为关键的是,其回归式架构仅支持输出一类最可能的构象,难以有效捕捉结合模式的多样性,尤其在结合构象高度灵活的PpI系统中,表现受到显著限制,且尚难满足大规模结构预测与虚拟筛选等实际应用需求。
RAPiDock架构
本研究提出了RAPiDock,一个专为蛋白–肽原子级对接设计的扩散生成模型(Diffusion Generative Model),旨在精确模拟肽分子在与蛋白结合过程中的空间构象变化,同时兼顾构象多样性与建模效率,满足蛋白-肽对接任务对精度和速度的双重要求。与传统依赖欧几里得空间的扩散方式不同,RAPiDock专为蛋白-肽结合建模任务设计了一套作用于四类结构自由度上的扩散过程,具体包括:整体平移、整体旋转、主链扭转角以及侧链扭转角。每类自由度均通过特定的扩散轨迹来建模其扰动与去噪过程,从而在确保生成结构物理合理性与构象多样性的基础上,有效压缩肽分子在高维构象空间中的采样范围,显著提升采样效率。为了充分建模肽分子的多尺度结构信息,RAPiDock引入了双尺度图结构:残基层图用于表示氨基酸残基间的全局几何构象与拓扑连接关系,原子层图则聚焦于原子级的空间排列与局部结构细节。两层图结构通过跨尺度信息融合机制实现特征交互,使模型在保留局部结构表达能力的同时具备较强的全局一致性与建模效率。在建模初始阶段,RAPiDock首先通过结构初始化模块,将肽序列转化为具有典型二级结构特征的初始三维构象。随后模型引入扩散过程,使结构进入无序、随机状态。随后,模型利用训练获得的反向去噪网络引导肽链从无序状态逐步演化回物理合理的有序构象,最终生成一系列物理合理、构象可信的结合模式。这一框架摆脱了传统对接方法对搜索算法与打分函数的强依赖,显著增强了蛋白–肽对接任务中的结构建模能力与多样性表达水平。

图1. RAPiDock模型架构图
RAPiDock在蛋白-肽对接准确性方面的表现
在蛋白–肽结合模式预测任务中,RAPiDock展现出卓越的结构预测准确性。在多个评估标准下,RAPiDock均优于当前主流的结构预测工具,包括AlphaFold3、AF2Multi以及ADCP。
在CAPRI-peptide评估体系的“可接受”标准下,RAPiDock在Top‑1层级已达到81.3%的成功率,Top‑25更是高达93.7%,较AF2Multi提升了13.4%。即便在更高标准的“高质量”评估下,RAPiDock的Top‑100成功率依然达到53.3%,较AF2Multi和ADCP分别提升了17.5%和29.8%,充分展现其在高精度构象预测方面的优势。值得强调的是,RAPiDock不仅能准确生成单一构象,还能在多个预测结果中提供兼具结构多样性与物理合理性的候选构象,显著增强了对真实结合状态的覆盖能力。研究还引入DockQ这一连续评分指标,对模型预测结构的整体质量进行系统性评估。评估结果显示,在多个评价层级中,RAPiDock均优于AlphaFold3、AF2Multi以及ADCP。在Top-1与Top-5层级下,RAPiDock分别有57.02%与61.84%的预测结果在DockQ得分上优于AlphaFold3。
进一步地,研究团队还系统评估了训练集与测试集之间的结构相似度对模型性能的影响。结果显示,即使在肽序列与蛋白结构与训练数据相似度较低的情况下,RAPiDock仍保持94.74%的预测成功率,而AlphaFold3在相同条件下仅为57.89%。这一结果表明,RAPiDock在低同源性样本上依然具备强大的结构采样与建模能力,展现出良好的泛化性能与鲁棒性。
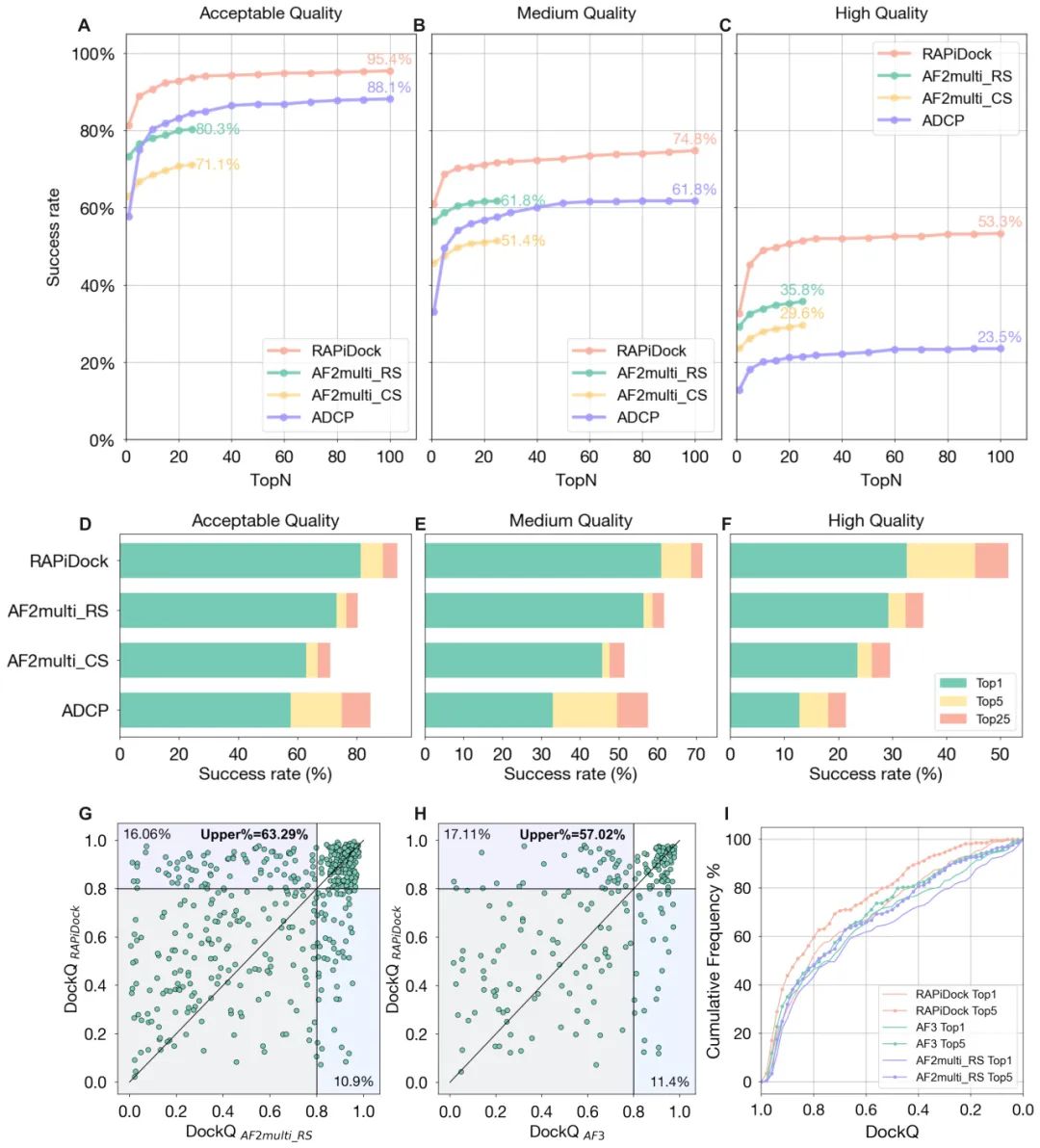
图2. RAPiDock在蛋白-肽对接准确性方面的表现
RAPiDock能够生成结构合理的多肽构象
在蛋白–肽对接任务中,所生成肽链结构能否符合真实的物理与几何规律,是衡量模型预测质量的关键标准之一。高质量的预测模型不仅需准确识别结合位置,还必须生成在构象上物理合理、统计分布可信的结构。为系统验证RAPiDock所生成多肽构象的合理性,研究团队对其输出的预测结构与对应晶体结构进行了对比分析。结果显示,RAPiDock输出的肽链在主链二面角的分布与晶体结构高度一致,KL散度均小于0.1,说明模型在构象层面高度复现了真实结构特征。同时,生成构象在Ramachandran图上的分布高度集中于统计允许区域,与实验结构保持高度一致。
此外,根据实验显示,初始二级结构的设置对RAPiDock最终预测结果几乎没有影响。这一特性表明,RAPiDock对初始构象依赖性低,具备出色的自我收敛性与结构泛化能力,赋予其在处理多类型肽分子时更高的灵活性与稳定性。
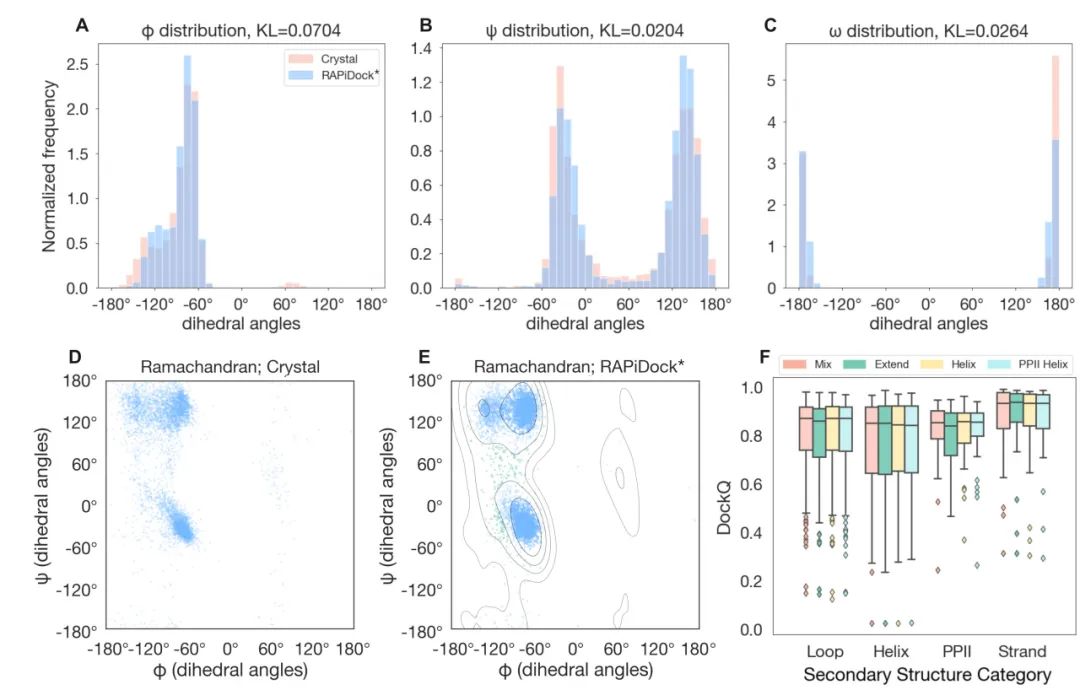
图3. RAPiDock生成构象的结构合理性分析
RAPiDock精准还原PLK1-PBD结合模式
Polo样激酶1(PLK1)在细胞有丝分裂过程中发挥关键调控功能,其异常表达已被证实与多种肿瘤的发生密切相关。其中,PLK1的Polo-box结构域(PLK1‑PBD)为典型的浅表型结合口袋,天然适配肽类分子的结合,是开发肽类抑制剂的重要结构靶点。
多个PLK1‑PBD与肽复合物的晶体结构显示,不同肽分子的结合会引发结合口袋内侧链构象的微小变化,给结构预测提出更高的精度要求。为验证RAPiDock在该类结合模式中的表现,研究团队选取了两个具有代表性的复合物结构(PDB ID: 8JOY和4E67),用于评估其对PLK1‑PBD结合构象的还原能力。
实验结果表明,RAPiDock可准确捕捉如磷酸化苏氨酸等关键锚定残基的相互作用,并成功复现结合口袋内的关键侧链重排。在8JOY案例中,RAPiDock生成的Top-2构象已达到CAPRI-peptide的高质量标准,准确还原了肽链N端卷曲的结合构象;对于4E67复合物,RAPiDock的Top‑1构象准确再现了肽链沿结合口袋延展排列的结构特征,并成功捕捉到芳香堆积等关键非共价相互作用。
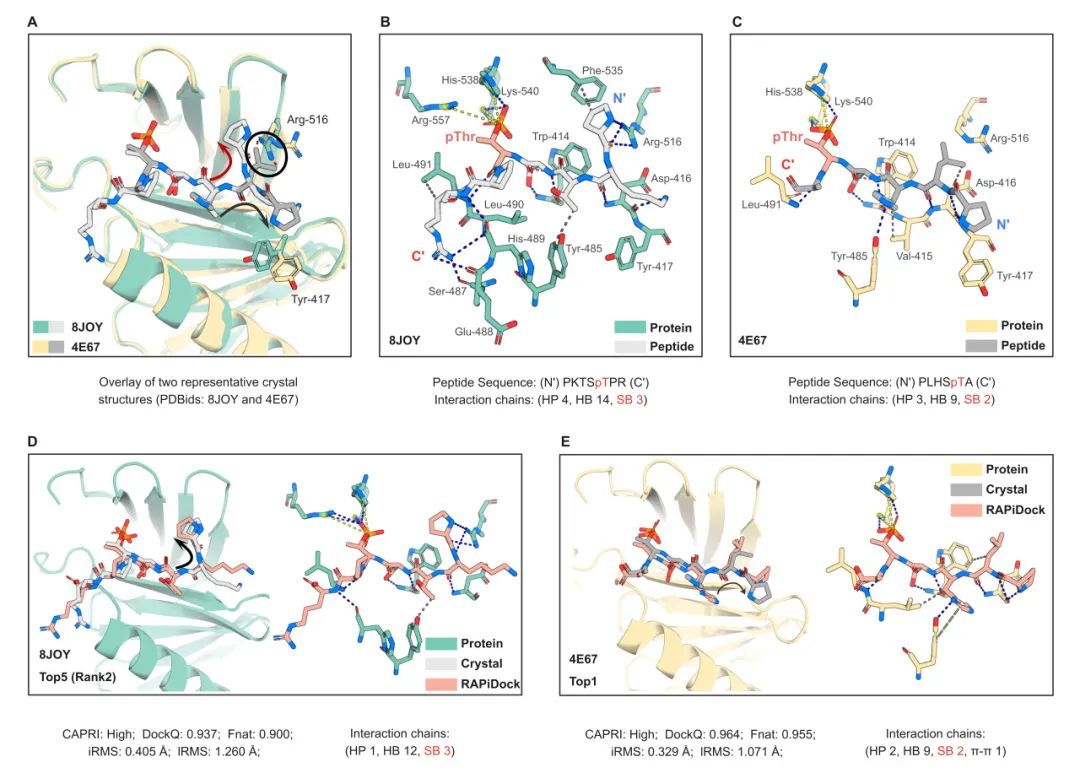
图4. RAPiDock精准还原PLK1-PBD结合模式
RAPiDock识别蛋白质中多个肽结合位点
在真实生物体系中,许多蛋白质不仅拥有一个肽结合位点,还可能存在多个结构差异显著的功能性结合口袋,这给结合模式的准确预测带来了更大挑战。在该类多结合位点识别任务中,RAPiDock展现出卓越的位点识别能力与构象判别精度。以核定位信号(Nuclear Localization Signal, NLS)受体Importin‑α为例,该蛋白包含一个主要结合口袋和一个次要结合口袋,构成典型的多位点结合体系。在全局对接模式下,RAPiDock成功识别出Importin‑α的两个结合口袋,并在前5个预测结构中同时捕捉到两个高质量构象:Top‑1构象准确命中次要口袋,Top‑5构象定位于主要口袋,均符合CAPRI‑peptide评估中的高质量标准。相比之下,AF2Multi仅识别出主要结合位点,且对应构象质量为中等水平;而传统方法如HPEPDOCK虽然也能识别两个口袋,但难以生成高质量预测结构,且结果排序准确性较差,实用价值受限。这些结果显示RAPiDock有望为多靶点蛋白的肽药设计与功能调控研究提供了高效且可靠的建模解决方案。
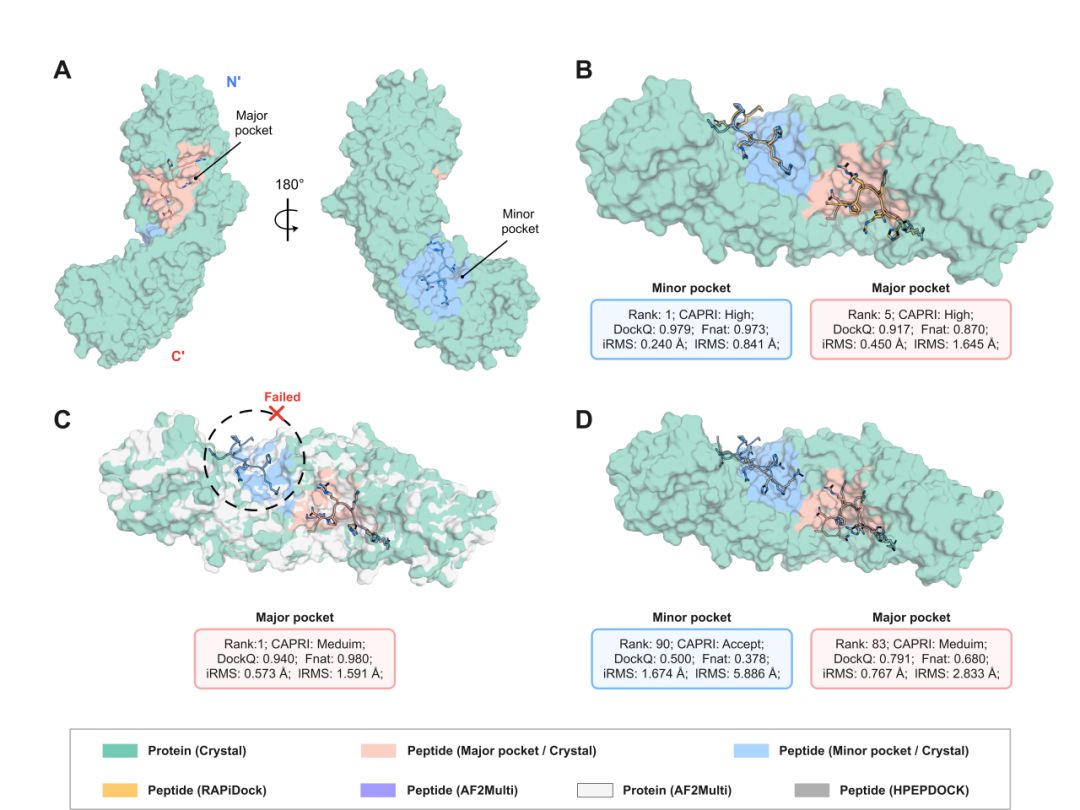
图5. RAPiDock能够识别蛋白质中多个肽结合位点
RAPiDock助力HLA体系的多肽虚拟筛选
肽-人类白细胞抗原(peptide-HLA, pHLA)体系是抗原呈递等免疫过程的核心组成部分,也是蛋白–肽对接任务中最具代表性和研究价值的体系之一。高效且精准地预测肽分子与HLA分子的结合模式,对于疫苗设计、免疫治疗以及个体化医疗等方向具有重要的应用价值。在pHLA系统的建模任务中,RAPiDock同样展现出出色的结构预测与结合模式重构能力。在多个HLA的结构预测任务中,RAPiDock实现了68.9%的Top‑1高质量预测成功率,显著优于AF2Multi与ADCP等主流方法。
更具实用意义的是,RAPiDock不仅具备结构构象预测能力,还展现出优异的虚拟筛选性能。在12种不同HLA亚型体系中,研究团队评估了RAPiDock预测构象的结合亲和力表现。结果显示,基于PyRosetta打分函数的评估中,RAPiDock生成构象的平均AUC‑ROC与AUC‑PRC分别为0.748和0.528,显著优于基于模板的肽分子预测方法(分别为0.673和0.419)。其中,在HLA‑B*57:01等关键等位基因体系中,RAPiDock的AUC‑ROC值接近0.9,显示出极高的筛选精度与显著的应用潜力。
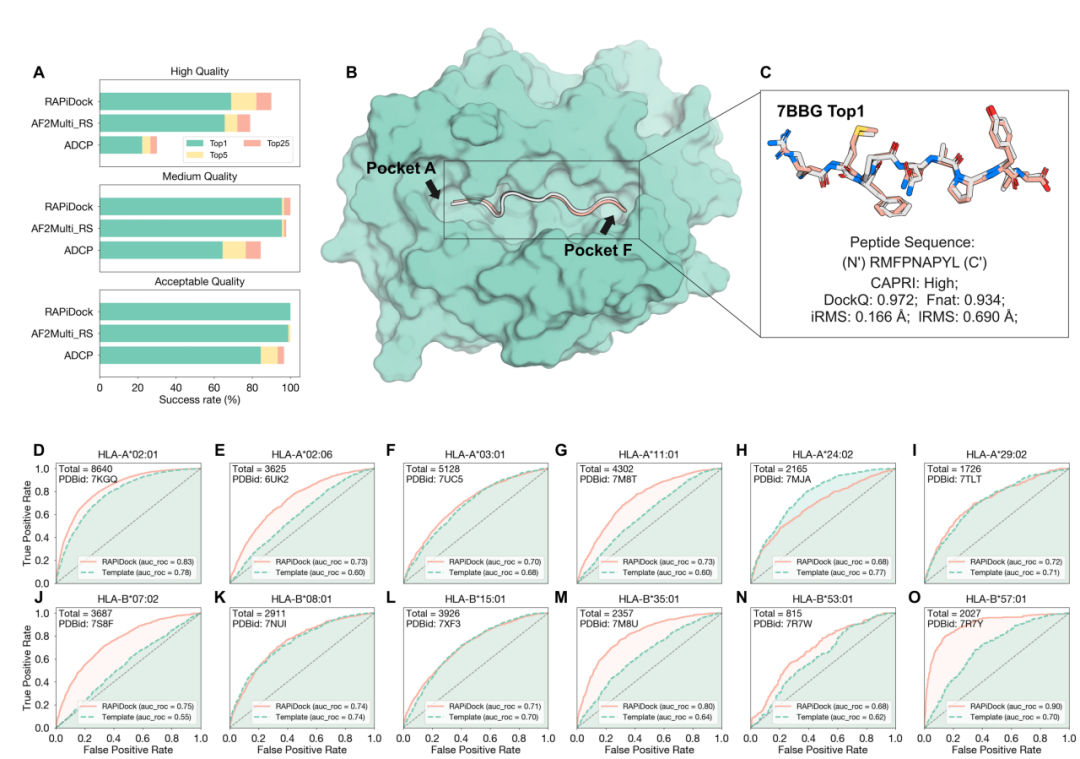
图6. RAPiDock助力HLA体系的多肽虚拟筛选
总结
RAPiDock的提出为PpI复合物的结构解析提供了全新的解决思路。作为一种基于扩散生成模型的对接工具,RAPiDock以其卓越精度、多样性建模能力和高计算效率,显著提升了蛋白–肽结合构象预测的可靠性。
在多种实际场景任务中,RAPiDock展现出优于主流方法的结构重构能力,能够准确还原复杂结合构象、识别多个潜在结合口袋,并支持翻译后修饰的自然建模。同时,其在pHLA结构预测与虚拟筛选任务中的出色表现,展示了其在免疫学、疫苗设计及个体化医疗等领域的广阔应用前景。
更为重要的是,RAPiDock不仅为蛋白–肽对接任务设定了新的精度与效率标准,也为结构生物学与计算药物设计的未来发展提供了方法论支持。随着高通量结构预测与筛选需求不断增长,RAPiDock有望成为推动肽类药物研发、蛋白功能调控以及新型靶点发现的重要技术支柱。
参考资料
Zhao, H., Zhang, O., Jiang, D. et al. Protein–peptide docking with a rational and accurate diffusion generative model. Nat Mach Intell (2025).
https://doi.org/10.1038/s42256-025-01077-9
内容中包含的图片若涉及版权问题,请及时与我们联系删除


评论
沙发等你来抢