
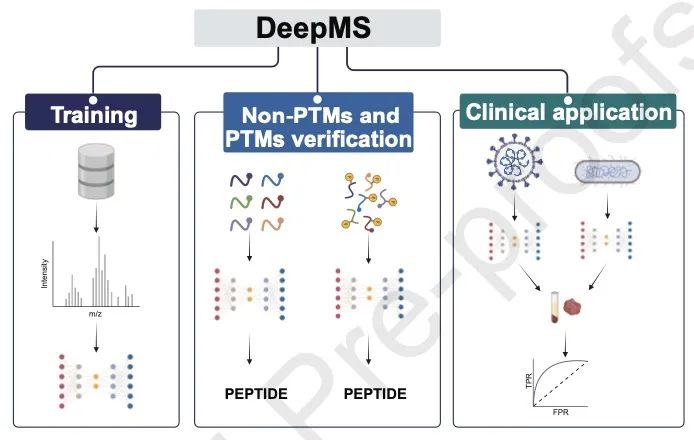
亮点
・开发了一种名为 DeepMS 的新型深度学习算法,能够实现端到端的质谱鉴定,处理速度超过每秒 3,000 个光谱,比传统工具快 20 倍以上。
・DeepMS 在大肠杆菌和 SARS-CoV-2 病原体检测中表现出卓越性能,敏感性和特异性均超过 98%,并能准确区分混合样本中的病原体。
・在未修饰肽段和翻译后修饰(PTM)鉴定中达到高达 94% 的准确率,优于其他深度学习模型,尤其在磷酸化和甲基化数据集上表现突出。
结果
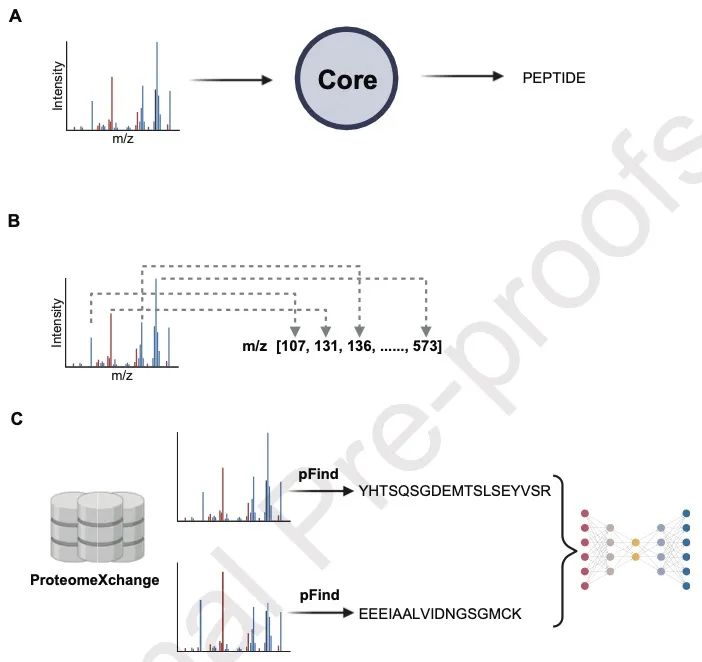
图1 DeepMS 算法的模型设计与训练流程
A:端到端 DeepMS 算法的框架设计。以质谱图为输入,通过深度学习架构输出肽段序列。
B:质谱特征提取。
C:深度学习算法的训练流程:使用 ProteomeXchange 数据库中的质谱数据,以 pFind 工具的初始鉴定结果作为正确序列,随后进行模型训练。
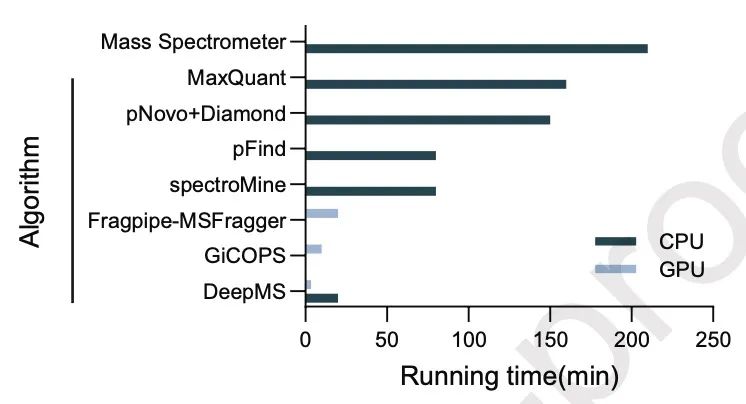
图2 DeepMS 与其他质谱鉴定工具在 CPU 和 GPU 平台上的耗时对比,包括 MaxQuant、pNovo + Diamond、SpectroMine、pFind、Fragpipe-MSFragger、GiCOPS。需注意,DeepMS 仅执行序列级鉴定,而其他工具可能包含蛋白质推断、FDR 控制和定量等额外处理步骤。图中同时标注了质谱仪生成该数据集的运行时间作为参考。
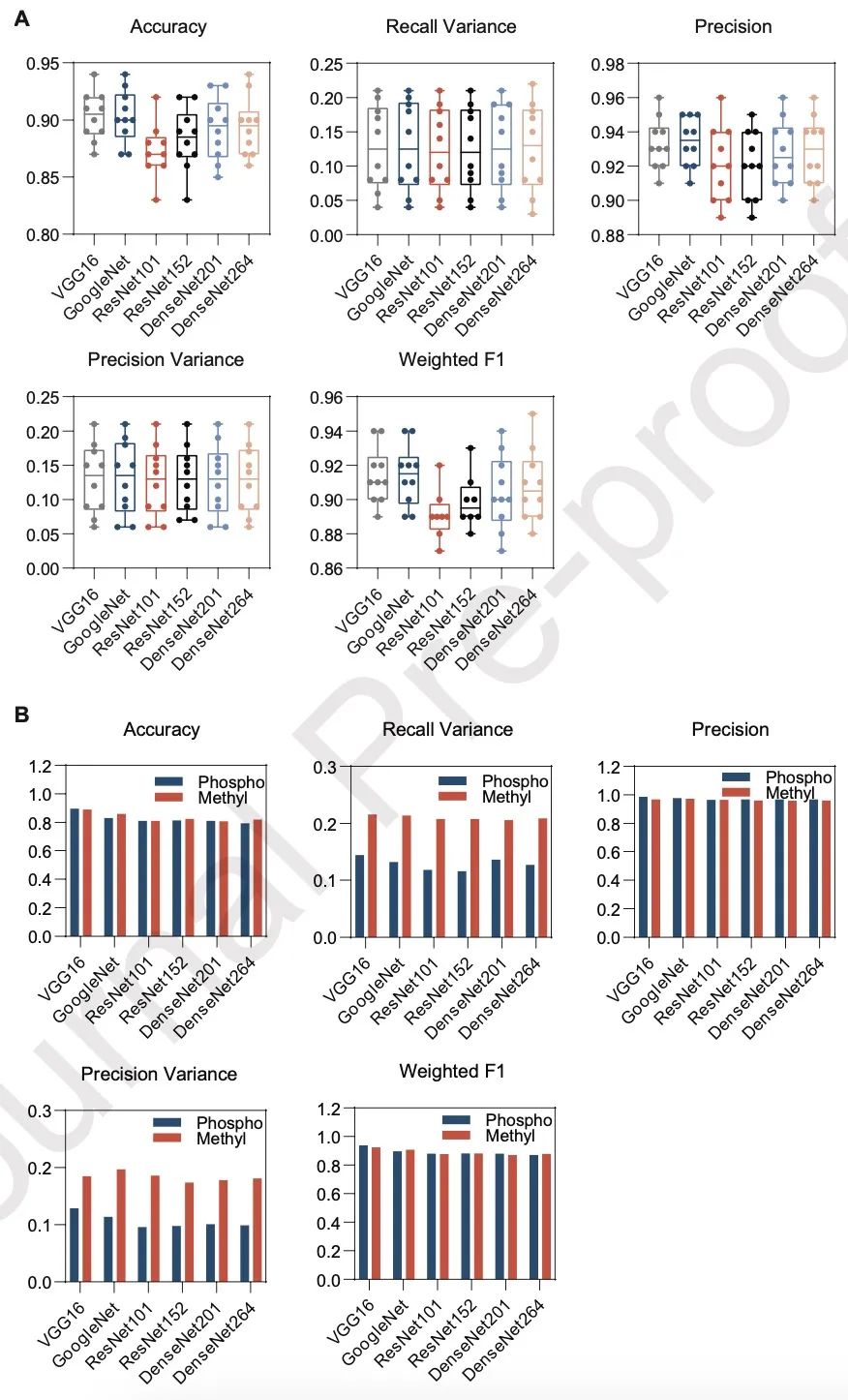
图3 六种深度学习模型在未修饰肽段数据集(A)和翻译后修饰富集肽段数据集(B,磷酸化和甲基化)中的验证结果。评估指标包括准确率、精确率、召回率方差、精确率方差和加权 F1 分数。
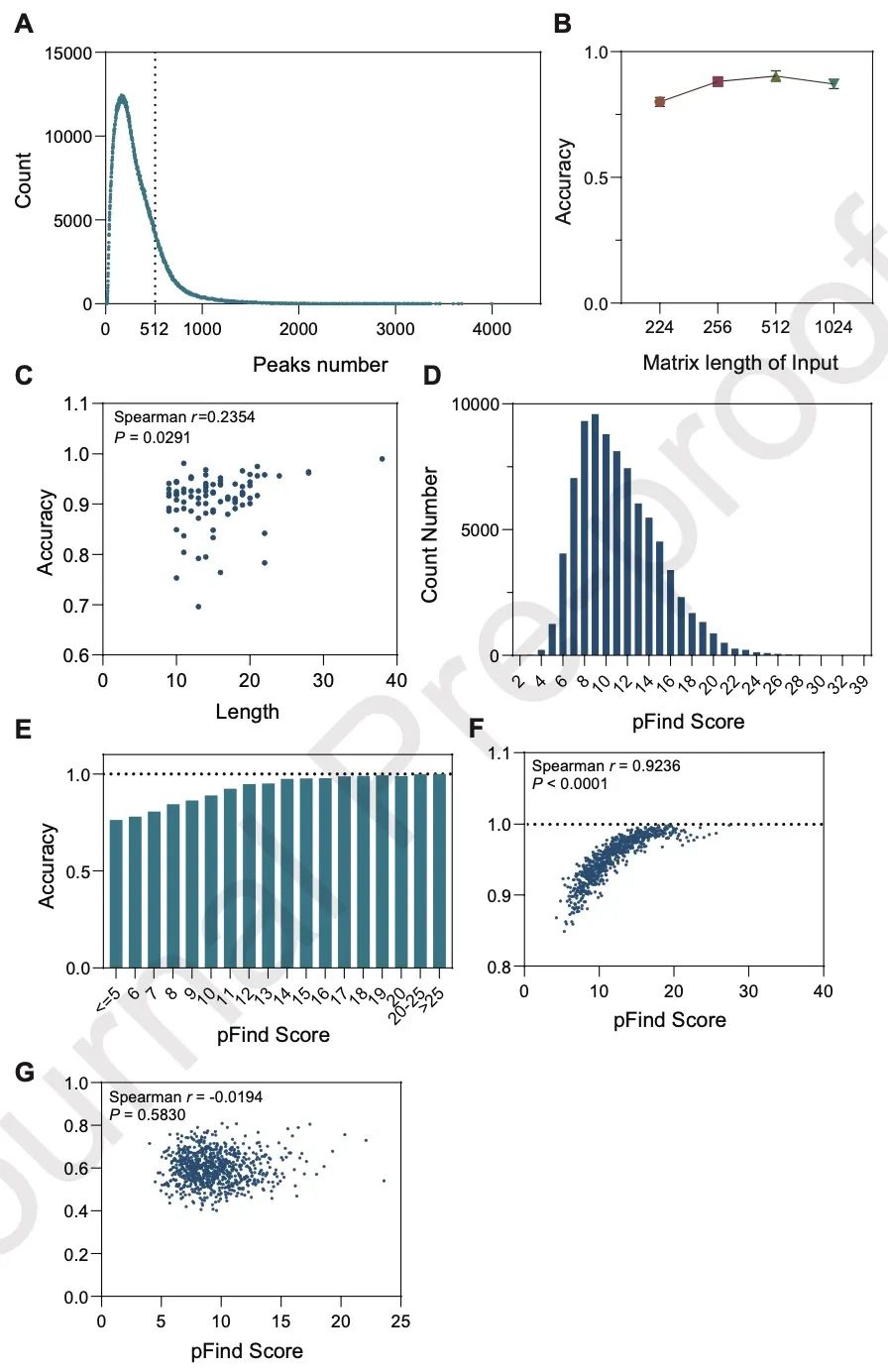
图4 质谱图质量与 DeepMS 准确率的关系
A:测试集中所有质谱图的峰数分布
B:输入特征矩阵长度与准确率的关系
C:肽段序列长度与准确率的关系
D:测试集中质谱图的 pFind 评分分布
E:预测准确率与 pFind 评分(匹配质量)的关系
F:预测正确的质谱图中,pFind 评分与预测概率的相关性
G:预测错误的质谱图中,pFind 评分与预测概率的相关性
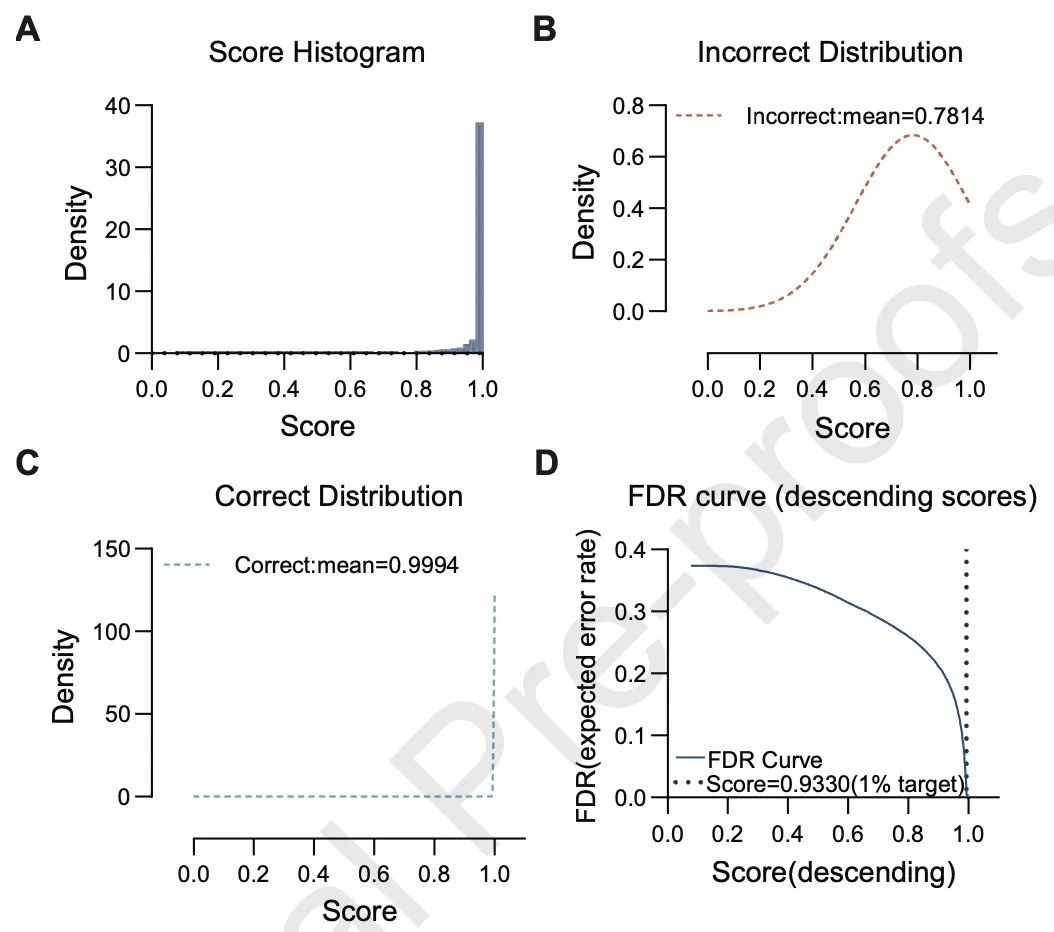
图5
A:DeepMS 所有预测结果的评分直方图
B:贝叶斯高斯混合模型(GMM)拟合的错误成分概率密度曲线
C:贝叶斯高斯混合模型(GMM)拟合的正确成分概率密度曲线
D:错误发现率(FDR)随评分变化的累积曲线。黑色虚线表示评分 = 0.9930 时对应的 1% FDR 阈值
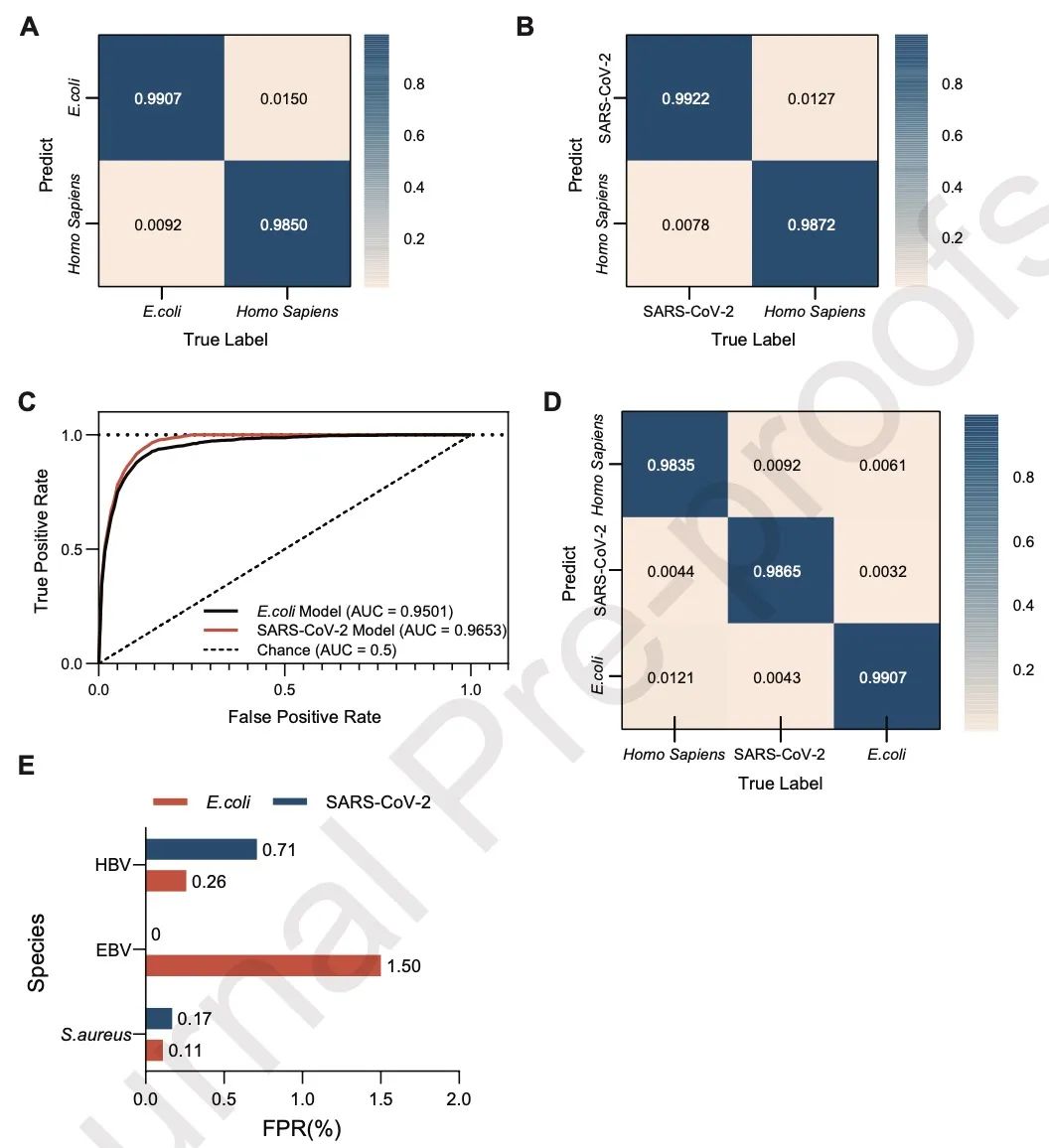
图6 DeepMS 算法在病原体阳性样本上的验证结果
A:大肠杆菌阳性样本预测结果的混淆矩阵
B:SARS-CoV-2 阳性样本预测结果的混淆矩阵
C:大肠杆菌和 SARS-CoV-2 阳性样本预测结果的 ROC-AUC 曲线
D:包含大肠杆菌、SARS-CoV-2 和人类质谱图的混合样本混淆矩阵
E:大肠杆菌模型和 SARS-CoV-2 模型在其他病原体样本(HBV、EBV 和金黄色葡萄球菌)上的假阳性结果
参考
J Mol Biol. 2025 May 29:169237. doi: 10.1016/j.jmb.2025.169237
注:AI辅助翻译,如有错误欢迎指出。请以复制粘贴,附上本公众号名片的方式转载此文。
End
内容中包含的图片若涉及版权问题,请及时与我们联系删除

评论
沙发等你来抢