
DRUGAI
药物-靶标相互作用预测是药物发现中的核心问题。传统基于结构的方法(如分子对接)虽然性能优异,但高度依赖精确的蛋白质三维结构。然而,许多关键靶点缺乏可靠的结构信息,极大限制了这些方法的应用。相比之下,基于序列的方法不依赖结构数据,但通常筛选性能不足、泛化能力有限,因此与基于结构的方法相比仍存在一定性能差距。
近日,浙江大学药学院盛荣团队在Journal of Chemical Information and Modeling期刊发表了一个基于序列的端到端的深度学习模型—HitScreen1,专门用于药物虚拟筛选场景。在严格去重的外部测试集 DUD-E 和 DEKOIS2.0 上,该模型仅通过序列信息就达到了与基于结构方法相当甚至略优的水平。这表明,即使缺乏蛋白质结构,仅凭序列信息也能实现有效的虚拟筛选。

HitScreen 的核心在于针对药物-靶标相互作用建模中数据与模型的不足提出了相应解决方案。如图1所示,在数据方面,团队提出了“条件标签反转”的数据增强策略,以缓解类不平衡、标注偏差与配体偏差问题。作者首先对蛋白-配体进行聚类,并将簇内所有阳性样本视为当前阳性。随后,从其他簇中为每个阳性样本挑选等量的候选配体作为“假定阴性”,以保持阳性与阴性样本的 1:1 平衡。同时,对挑选的阴性样本与当前阳性样本进行结构相似性限制,避免出现假阴性风险。这样的设计既抑制了模型过度依赖配体特征的“捷径”,又迫使其学习更真实的蛋白-配体互作模式。在模型方面,HitScreen 融合了多种预训练的蛋白语言模型(Ankh、ESM-2、ProtT5 等)与分子预训练模型(Uni-Mol),弥补了传统序列方法在空间结构表达上的不足。同时显式引入交叉注意力机制,在氨基酸残基与配体原子层面建立双向耦合,避免编码结果只依赖全局独立表示,更直接地学习局部相互作用。
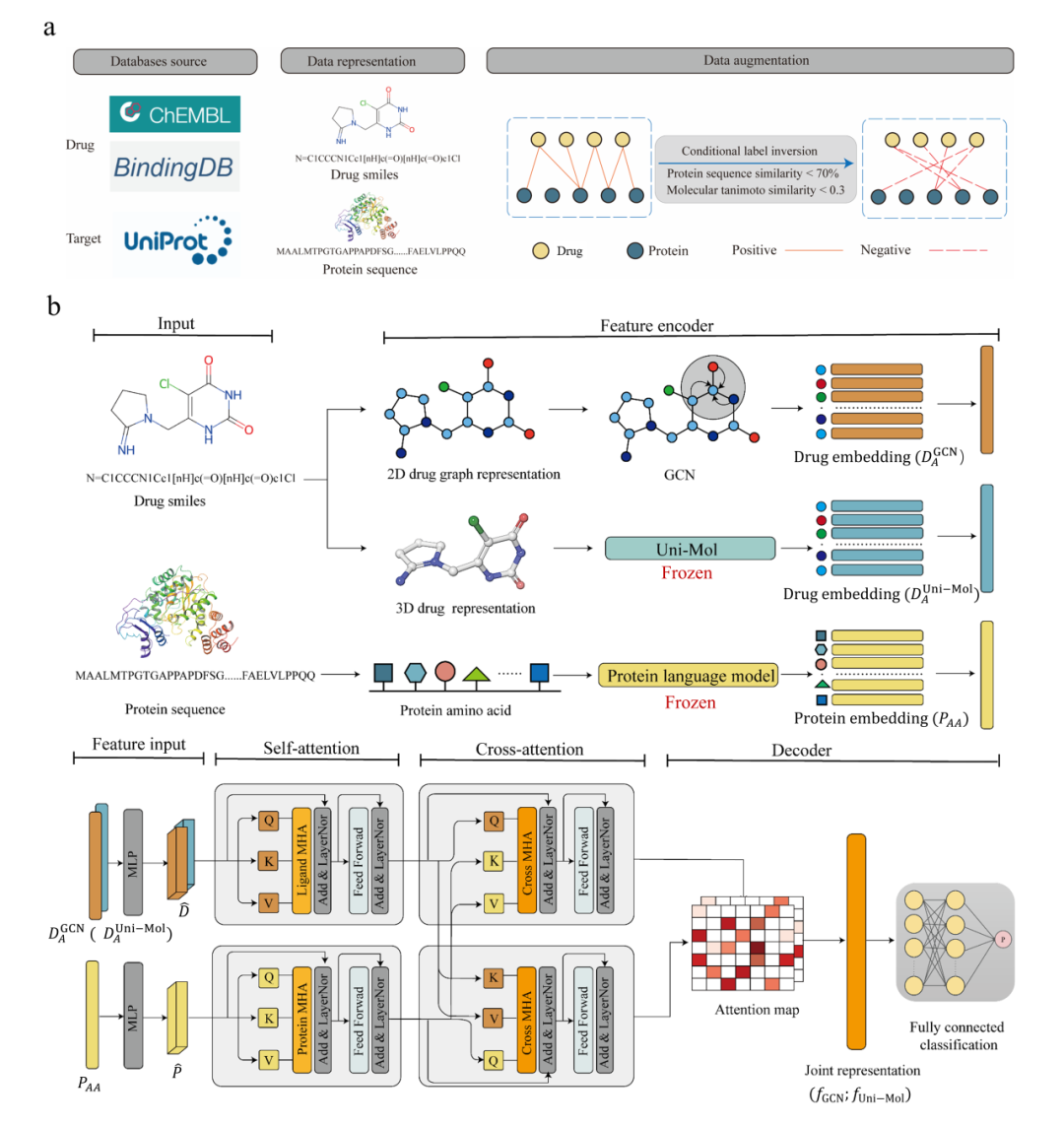
图1. (a)数据集的构建及数据增强策略。(b)HitScreen 模型框架。
数据增强策略的有效性验证
为了验证数据增强策略的有效性,作者在 Bai 等人2构建的 BindingDB 低偏差版本和 DrugBAN 模型上,对比了四种不同方法:1. 条件标签反转(本文方法);2. 基于网络的负样本;3. 生成模型 DeepCoy;4. 随机负样本。结果如图2所示,不同策略均能提升模型性能,其中条件标签反转表现最优。这一结果既凸显了数据增强的重要性,也验证了该方法的通用性。
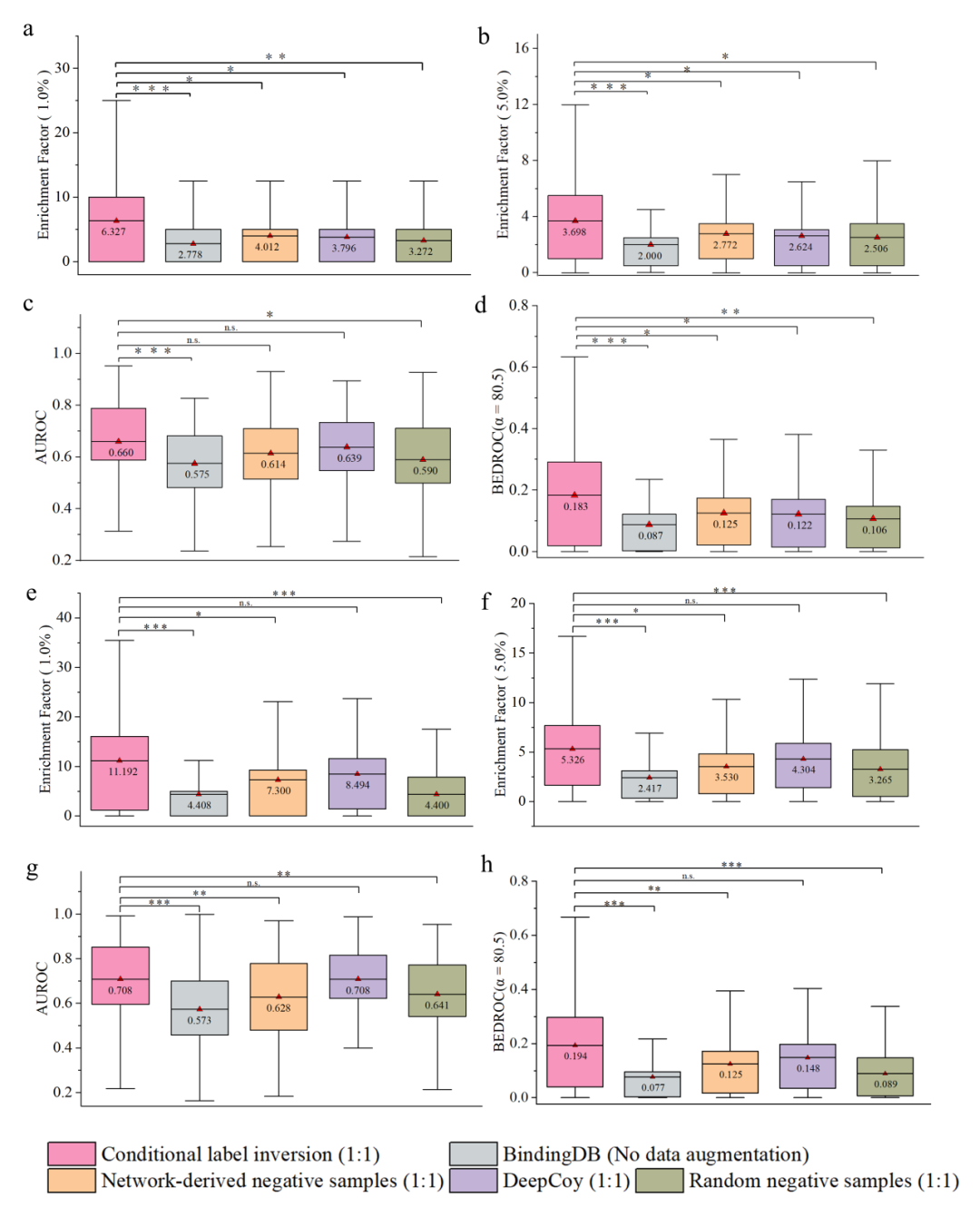
图 2. DrugBAN 模型在 BindingDB 数据集上对不同数据增强方法进行比较。(a-d)不同数据增强方法在 DEKOIS2.0 数据集上的表现。(e-h)不同数据增强方法在 DUD-E 数据集上的表现。
蛋白语言模型的影响
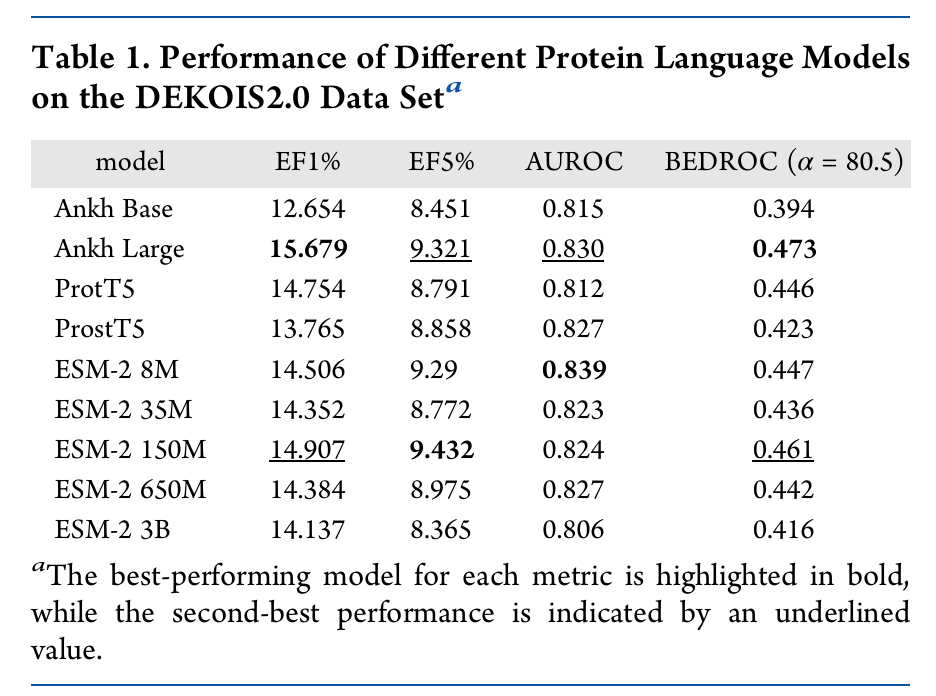
在蛋白语言模型的选择上,作者在 HitScreen 框架中评估了 9 种不同的模型,均在 ChEMBL33 数据集上训练,并在 DEKOIS2.0上测试。结果如表1所示,不同的蛋白语言表征确实会存在性能差异,但整体差距不大。
数据泄露风险和泛化性评估
在虚拟筛选场景中,数据泄露风险主要来自两个方面:一是部分活性分子可能同时对多个相关靶点具有活性;二是属于同一家族或亚家族的蛋白质往往共享结构或功能特征。前者可能导致分子在不同靶点间的信息传递,后者则可能使模型在同源蛋白之间泄露特征。为了严格评估 HitScreen 是否受到这些风险的影响,作者选取了三种表现较好的蛋白语言模型(Ankh Large、ProtT5、ESM-150M),并设定了三类逐步严格的去重标准:首先,从训练集中移除所有出现在测试集的靶点-配体对及活性分子;其次,删除与相同 UniProt 靶点相关的数据;最后,根据序列相似性划分,严格排除在 30% 阈值下相似的同源蛋白。结果如图3所示,随着去重标准逐步严格,三种模型的性能均出现下降,说明数据泄露风险确实存在。但值得注意的是,基于 Ankh Large 的模型下降幅度最小,显示出相对较强的泛化能力。
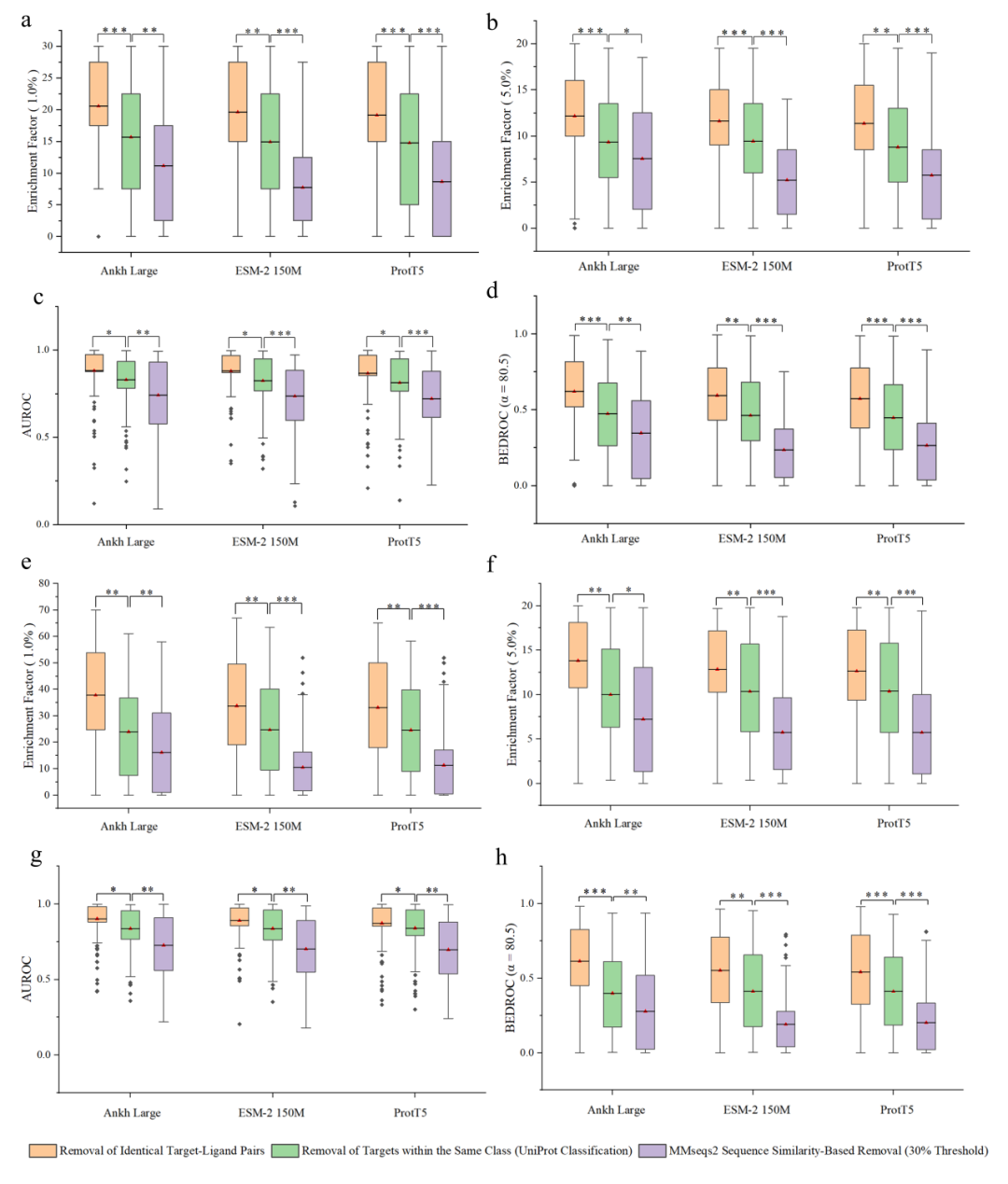
图3. 对三种蛋白质语言模型在不同去重标准下的泛化性能评估。(a-d)DEKOIS2.0 数据集。(e-h)DUD-E数据集。
HitScreen达到了基于结构方法的筛选性能
为了比较HitScreen的筛选能力,作者将HitScreen(Ankh Large)与27种现有方法在DUD-E数据集上进行比较,包括传统分子对接(Glide SP)、基于结构的深度学习方法(RTMScore、EquiScore等)、基于序列的方法(TransformerCPI2.0、DrugBAN、PSICHIC等)以及早期的机器学习方法。结果如图4所示,现有的序列方法通常表现不如结构方法,只有 TransformerCPI2.0 和 DrugBAN 在部分指标上达到中等水平。而 HitScreen 在所有关键指标上均表现优异,即使在靶标去重的情况下,依然能够与基于结构的方法相当甚至略优。
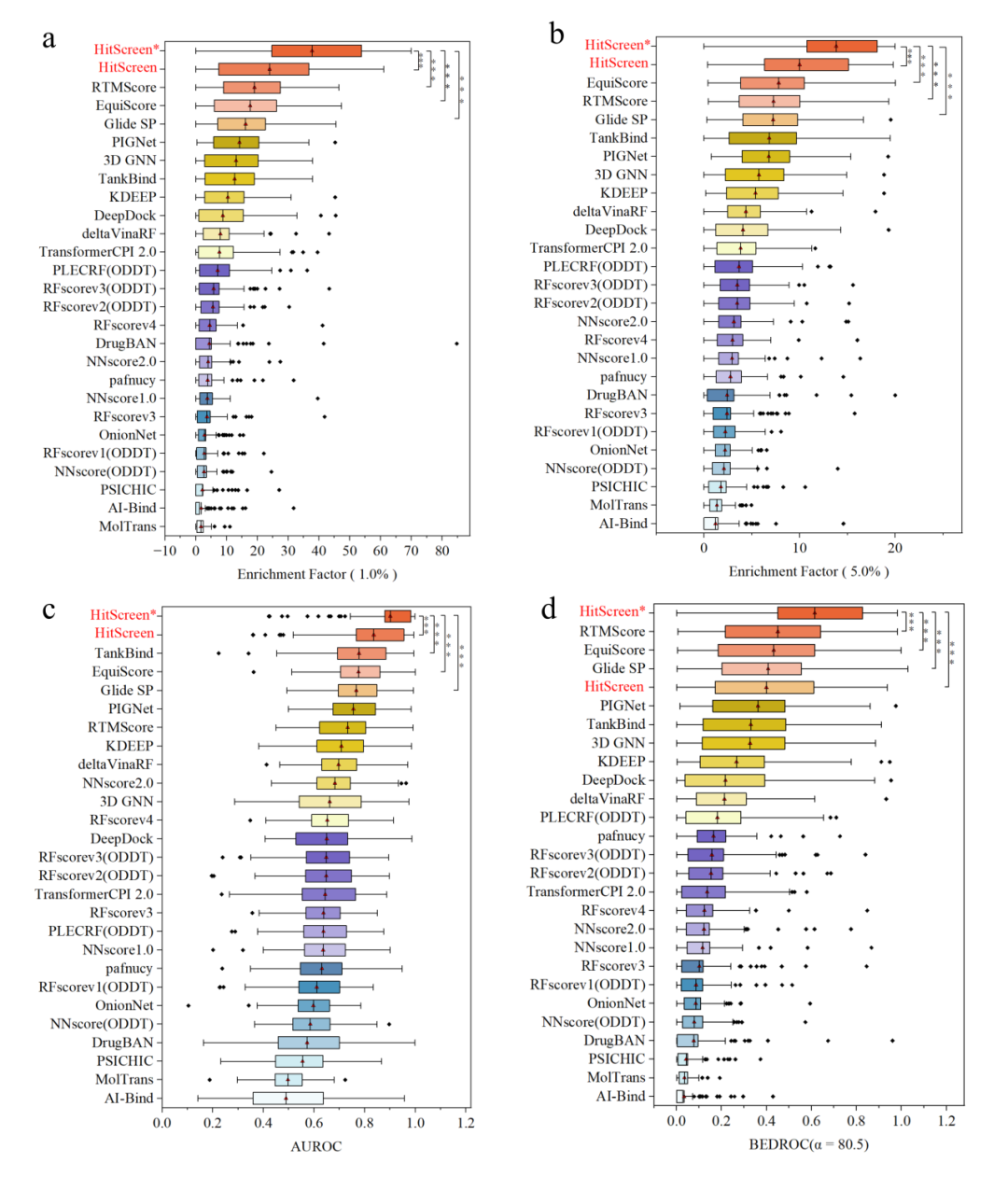
图4. 27 种打分函数在 DUD-E 数据集上的评估(HitScreen*为靶标-配体对去重,HitScreen为UniProt靶标去重)。
消融实验:各模块的贡献分析
为了量化各模块贡献,作者进行了系统消融研究,包括数据增强策略、蛋白质语言模型、Uni-Mol、GCN与特征交互模块五个要素。以Ankh Large为例,在DEKOIS2.0和DUD-E上的结果如图5所示,不使用数据增强策略会导致性能下降;以CNN替代蛋白质语言模型编码也会降低筛选能力;将自注意力和交叉注意力替换为MLP,在缺乏显式互作建模时,性能也会下滑;移除Uni-Mol嵌入或GCN特征性能都会下降。值得注意的是,当移除数据增强、蛋白质语言模型表征或特征交互模块会造成筛选性能的显著性下降,说明这些模块的最为关键。
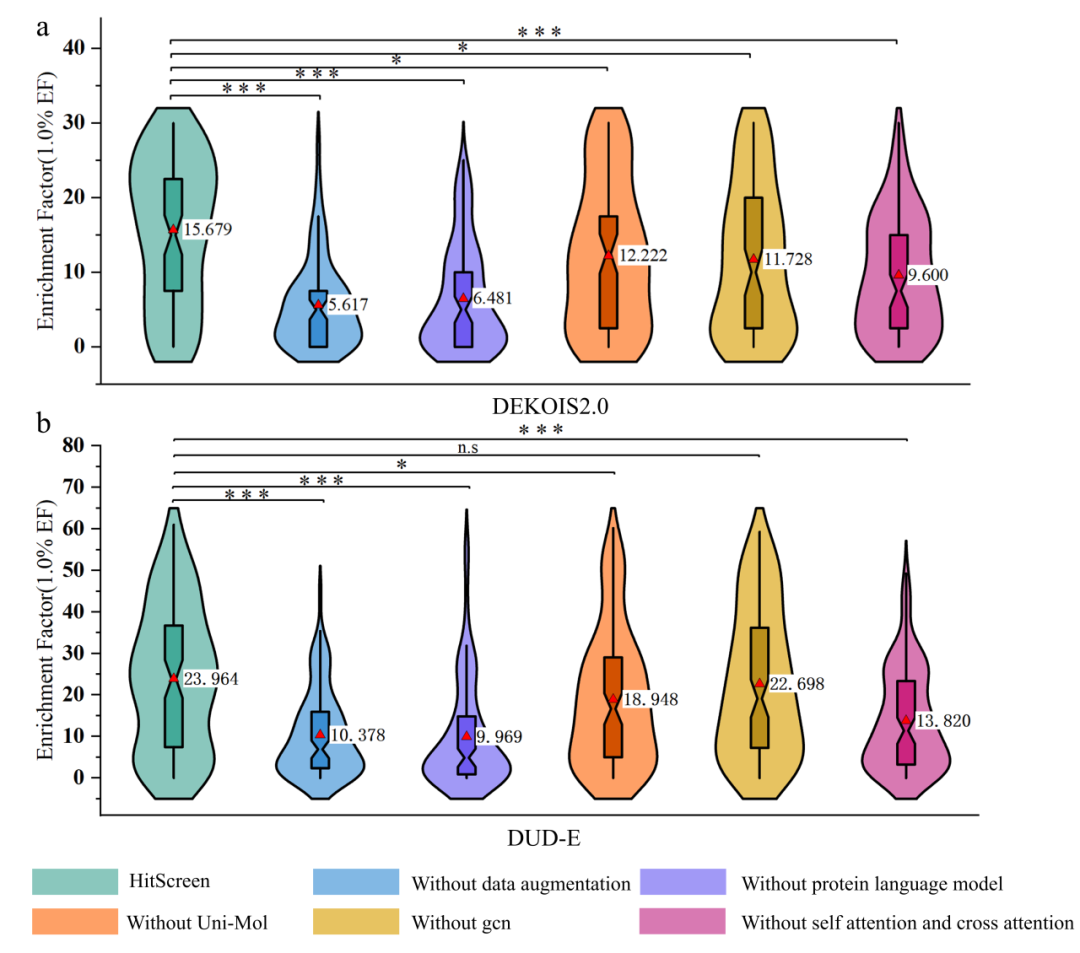
图5. HitScreen的消融实验。
可解释性分析:注意力机制的可视化
在可解释性方面,作者以 JNK2 蛋白复合物(PDB: 3NPC)为案例,结果如图6所示,展示了模型注意力热图与已知氢键互作之间的对齐关系。这表明即使没有显式输入三维结构或结合口袋位置,模型在数据驱动的学习过程中也能识别关键的原子-残基层面相互作用,这为理性药物设计提供了可信的线索。
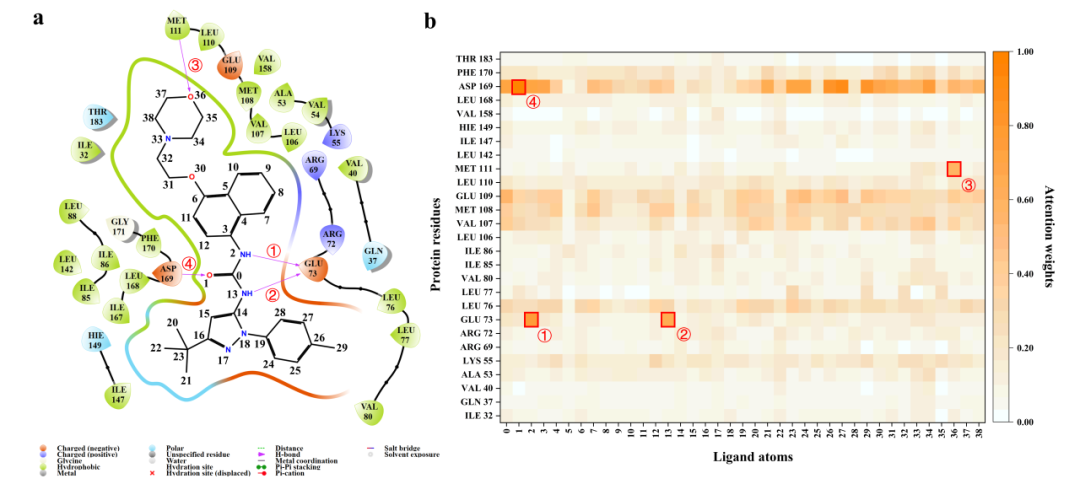
图6. HitScreen的可解释性分析。
总结与展望
综上,HitScreen 是一个端到端的基于序列药物-靶标相互作用预测框架,适用于药物的虚拟筛选场景。在数据层面,它通过“条件标签反转”机制有效缓解了数据不均衡与偏差问题;在模型层面,它整合了分子与蛋白的多重预训练表征,并显式建模局部相互作用。在多项严格评测中,HitScreen 的表现优于其它基于序列的方法,并可与先进的基于结构的方法相媲美。消融实验强调了数据增强、蛋白语言模型和交互模块的关键作用;注意力可视化则进一步表明,模型能够捕捉与生物互作一致的可解释模式。
展望未来,基于序列的药物-靶标预测有望进一步加深对药物靶标相互作用的理解,并加速药物开发。HitScreen 的研究证明,仅凭序列信息也能获得有价值的生物学见解,为药物发现提供了一种更灵活且可扩展的解决方案。
浙江大学博士研究生陈庚、廖金标、俞言真为本文的共同第一作者。本研究的源代码和数据可在 GitHub 获取: https://github.com/chengeng17/HitScreen.
参考资料
(1) Chen, G.; Liao, J.; Yu, Y.; Le, K.; Zhao, H.; Qin, Y.; Cai, L.; Sheng, R. HitScreen: A Sequence-Based Drug Virtual Screening Approach Using Data Augmentation and Protein Language Models. J. Chem. Inf. Model. 2025.
(2) Bai, P.; Miljković, F.; John, B.; Lu, H. Interpretable bilinear attention network with domain adaptation improves drug–target prediction. Nat. Mach. Intell 2023, 5, 126-136.
内容中包含的图片若涉及版权问题,请及时与我们联系删除

评论
沙发等你来抢