
DRUGONE
蛋白质–蛋白质相互作用(PPI)对生物功能至关重要。尽管基于协同进化分析和深度学习的蛋白质结构预测方法已在细菌和酵母中实现了大规模互作预测,但在更复杂的人类蛋白质组中应用仍然有限。研究人员通过两个关键创新克服了这一挑战:其一,利用来自30 PB未组装基因组数据的更深层多序列比对(MSA),增强了进化信号;其二,开发了基于2亿个预测结构的域–域互作(DDI)数据集训练的新型深度学习网络。研究人员系统性地筛选了2亿对人类蛋白质,最终预测出17,849个互作,预期精确度达90%,其中3,631个为先前实验未发现的新互作。这些预测的三维结构模型为探索蛋白功能及人类疾病机制提供了新的假设。

检测蛋白互作和解析蛋白复合物三维结构是理解蛋白功能的核心任务。实验手段如酵母双杂交(Y2H)和亲和纯化质谱(APMS)虽有重要贡献,但常受限于假阳性与假阴性率高、以及非生理条件下的检测。现有数据库虽记录了数十万互作,但一致性很低,仅有不到4000对PPI在多个数据库中同时被认为可信。
计算方法通过同源性、界面质量和功能关联来辅助互作预测。协同进化分析结合结构预测(如AlphaFold、RoseTTAFold)已在细菌、酵母中开展了蛋白质组规模的筛查,但在人体中仍受制于计算规模和有限的动物基因组数据。研究人员此前开发的轻量级深度学习网络在人类互作预测中精度有限,而AlphaFold2虽精度更高,但计算开销过大,难以应用于蛋白质组范围。
结果
基因组序列挖掘与进化信号增强
研究人员整合了超过2万种真核生物的基因组与转录组数据,构建了更深层的omicMSA,显著扩展了物种覆盖度,进而提高了检测协同进化信号的能力。与传统UniRef或宏基因组序列相比,omicMSA在区分真实互作与随机配对时表现更优。
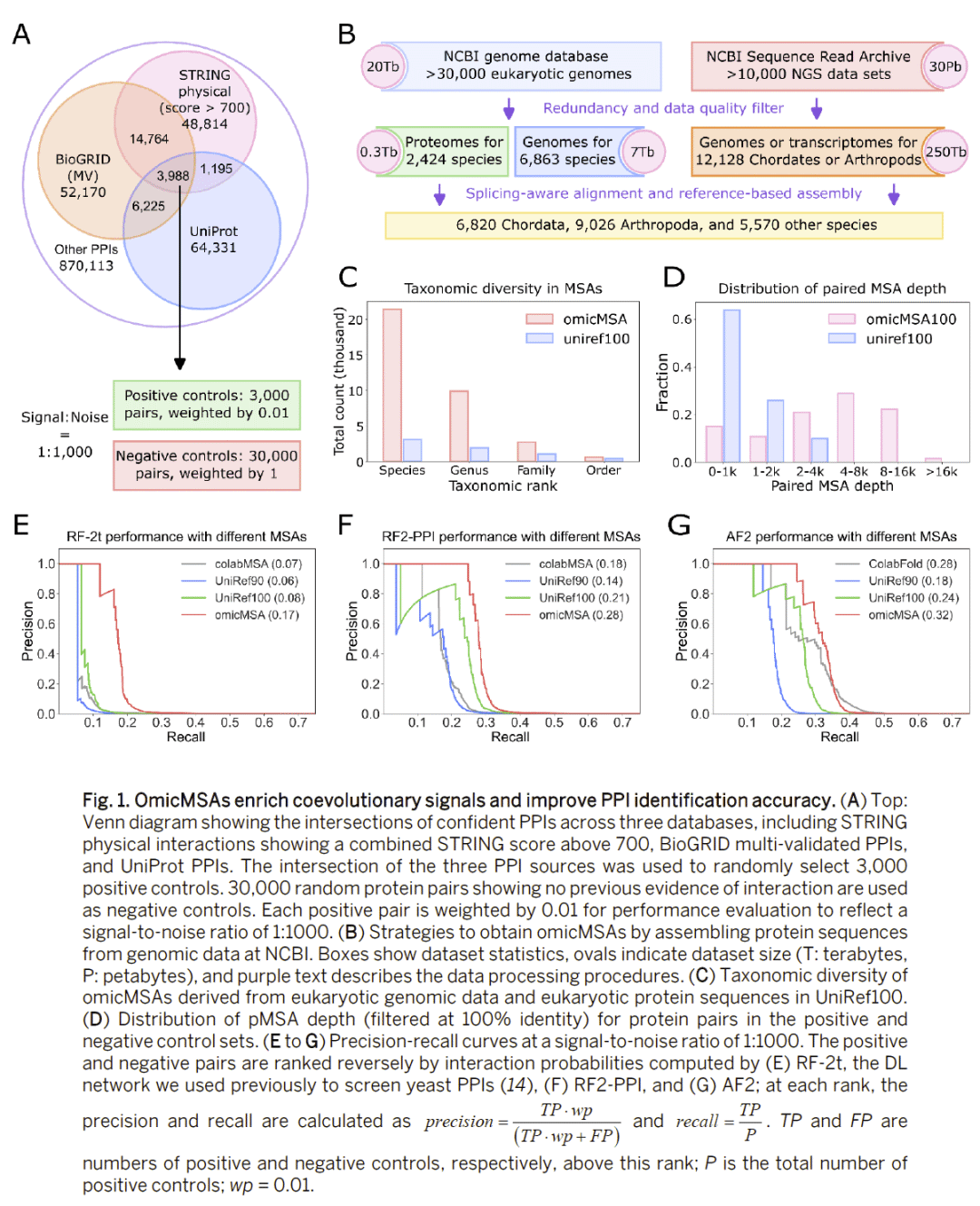
RF2-PPI:快速互作预测深度学习网络
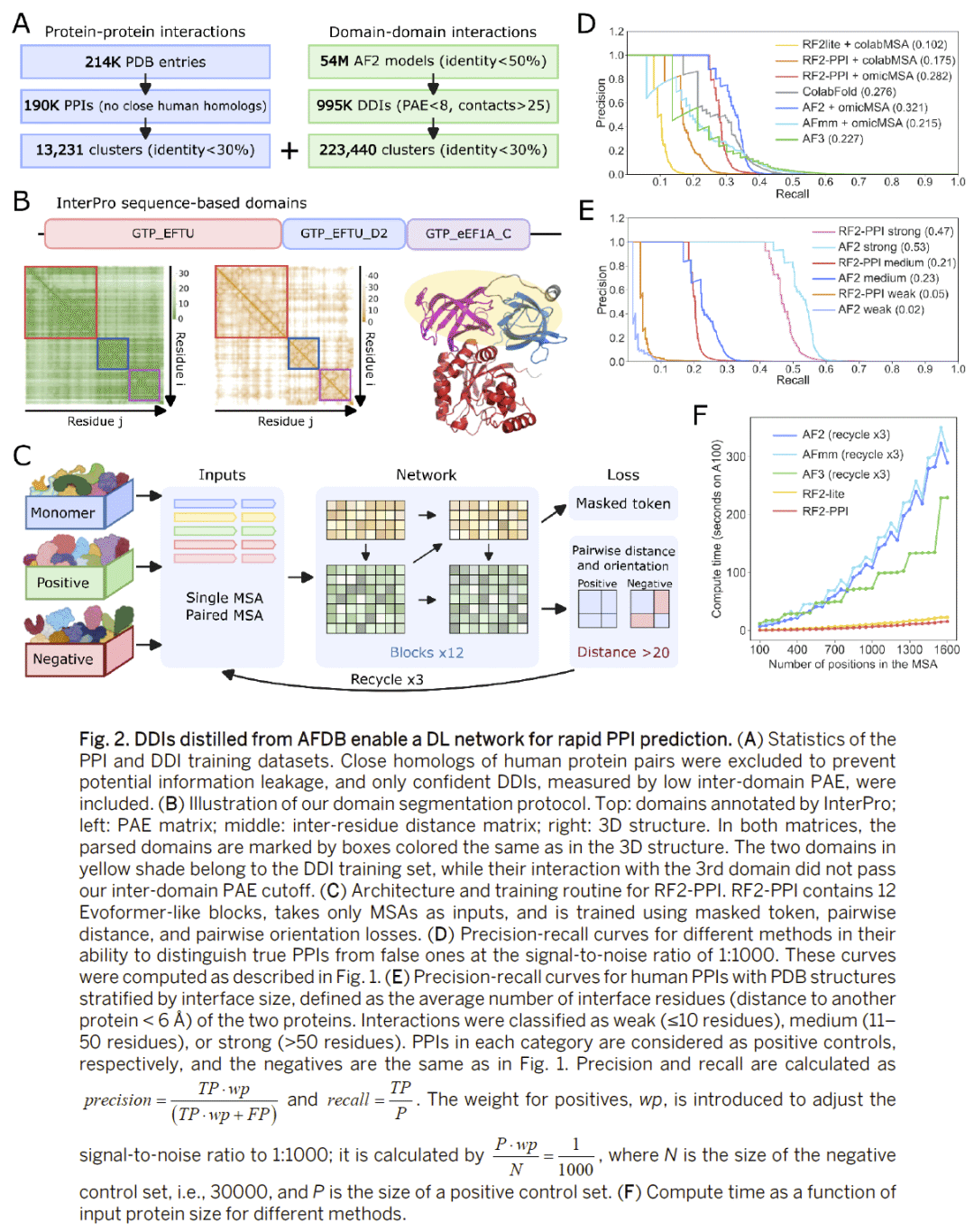
研究人员基于RoseTTAFold2架构,结合PDB的实验性PPI与从AlphaFold数据库提取的高置信度DDI,训练了一个大规模网络(RF2-PPI)。这一策略极大增加了训练数据规模,使其性能超越以往轻量化方法。与AlphaFold3或AF-multimer相比,RF2-PPI在区分真实互作和非互作方面更为准确,尤其在弱互作预测中具优势,同时计算效率显著提升。
人类蛋白质组范围内的互作预测
研究人员利用RF2-PPI和AlphaFold2,对19,528个人类蛋白(约1.91亿对配对)进行了筛选。通过分层过滤(直接耦合分析DCA、RF2-PPI评分、AlphaFold结构建模),最终在严格条件下预测出17,849个高置信度PPI(90%精度),覆盖人类互作组的约8–22%。
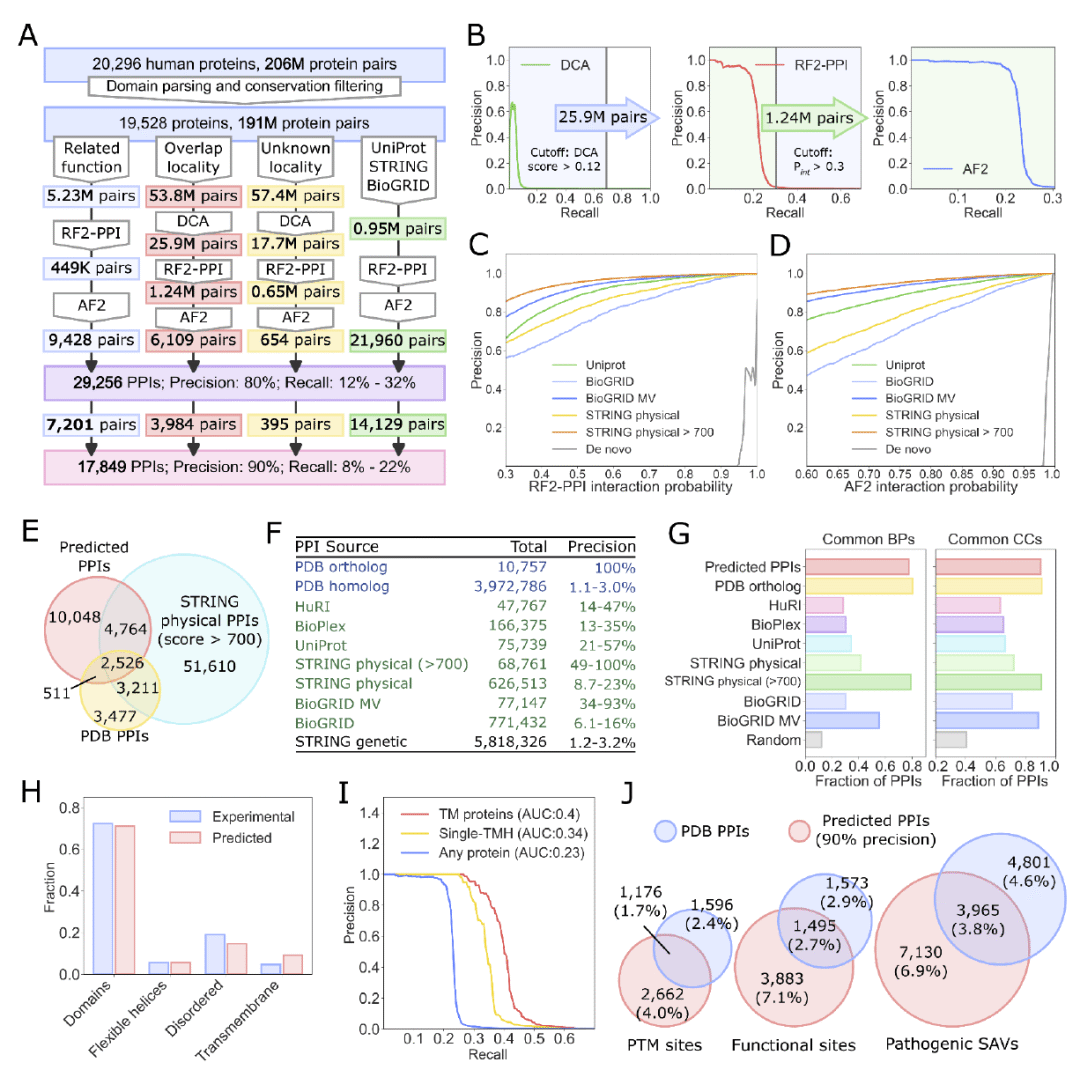
图 3. 蛋白质组范围的 PPI 筛选流程与统计。
与知识库整合与功能洞察
预测结果与现有数据库对比发现,研究人员的预测在功能一致性和亚细胞定位一致性上优于大部分已有数据集。结构建模显示,大部分互作由结构域介导,但也涵盖部分跨膜蛋白互作。值得注意的是,约15%的疾病相关单点突变可映射至预测互作界面,揭示了疾病可能的分子机制。
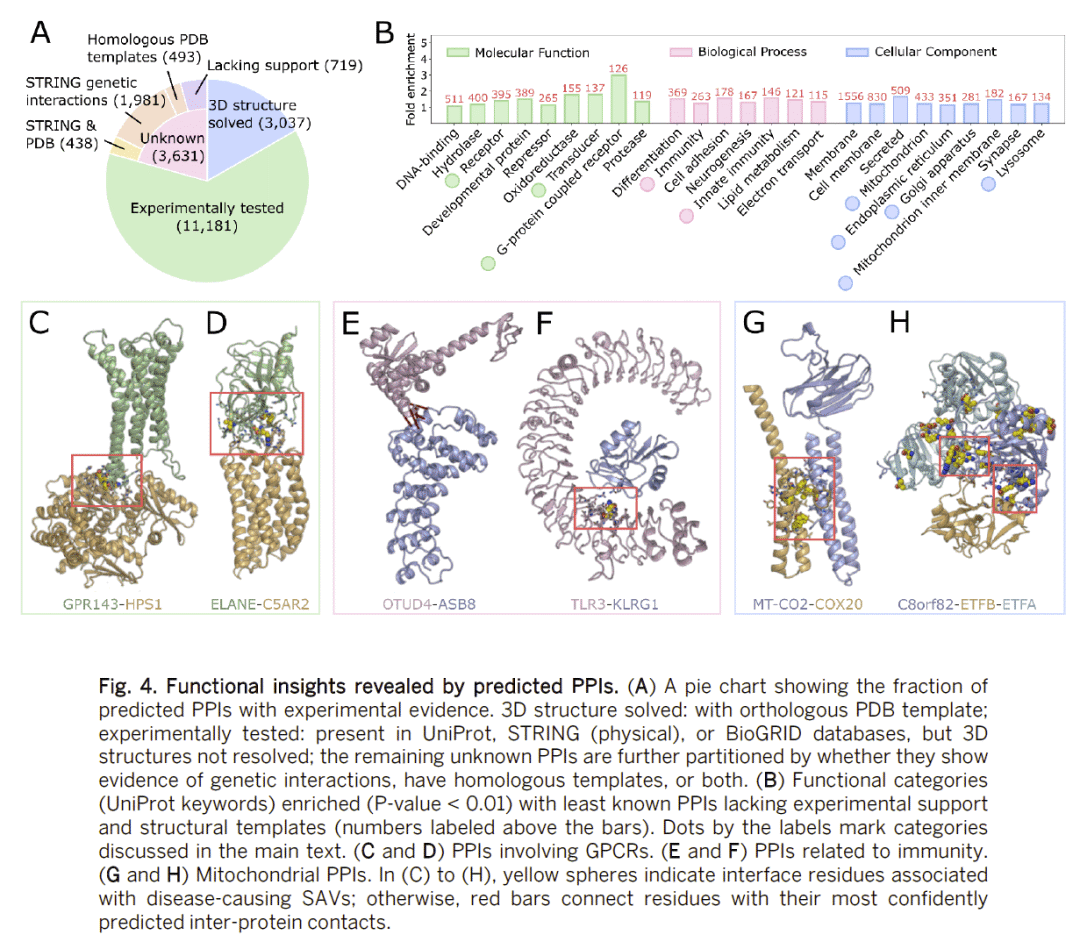
复合物组装与新增亚基预测
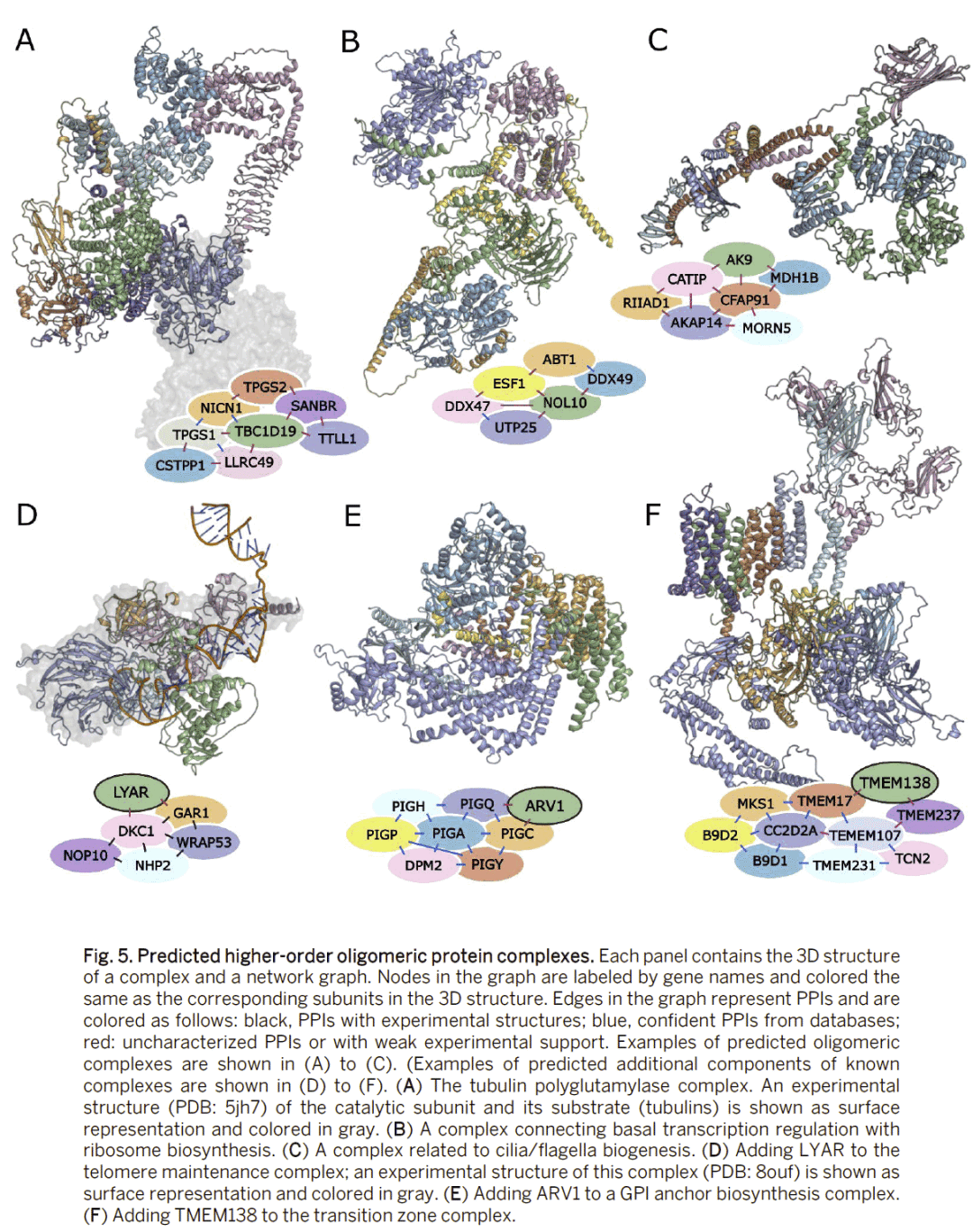
基于预测的互作网络,研究人员鉴定出数百个潜在多蛋白复合物,并为部分已知复合物预测了额外的亚基。例如,微管相关酶复合物和端粒维护复合物都被补充了新的组成成分。
讨论
研究人员通过结合更深的进化信号和更大规模的训练集,提出了一种在蛋白质组范围内预测人类PPI的有效方法,显著扩展了高置信度互作的覆盖率。这些预测不仅补充了现有实验数据的空白,还为功能未知蛋白的研究提供了新的切入点。特别是在跨膜蛋白和免疫相关互作方面,本研究的预测具有重要的生物学与医学意义。
尽管如此,预测仍受限于弱互作和瞬时互作的识别能力不足,尤其是由无序区介导的互作。未来,随着更多基因组数据和结构数据的积累,以及深度学习方法的不断发展,这一框架有望更全面地描绘人类三维互作组,加速对疾病机制和潜在药物靶点的理解。
整理 | DrugOne团队
参考资料
Jing Zhang et al. ,Predicting protein-protein interactions in the human proteome.Science0,eadt1630
DOI:10.1126/science.adt1630

内容为【DrugOne】公众号原创|转载请注明来源
内容中包含的图片若涉及版权问题,请及时与我们联系删除

评论
沙发等你来抢