
人工智能正以快速的步伐改变药物发现中的虚拟筛选,通过利用日益增长的实验数据并扩大其可扩展性,这些创新有望提升基于配体的虚拟筛选(LBVS)方法和基于结构的虚拟筛选(SBVS)方法的效率和精确性。然而,数据整理、新模型的严格且前瞻性的验证、以及与实验方法的高效整合,仍是实现人工智能在药物发现中全部潜力的关键挑战。
渥太华大学研究团队于2025年5月25日在《Expert Opinion on Drug Discovery》上发表文章,题为“On the application of artificial intelligence in virtual screening”。
文章旨在阐述人工智能如何通过SBVS和LBVS的成功案例,加速并提升治疗性药物发现的早期阶段,同时揭示正在出现的关键局限性,并提供对未来计算机辅助药物发现发展格局的概览。

背景
药物发现是一项极其复杂的任务,耗时且昂贵,通常需要十年以上的时间和超过20亿美元的投入,才能将一种小分子药物推向市场。虚拟筛选(VS)可以探索规模庞大的化学空间,涉及数十亿个化合物,从而能够选择潜在感兴趣的分子,并探索药物化学空间中前所未有的领域。这种方法不仅提高了发现新颖且高效配体的可能性,还能促进具有更优理化性质候选物的发现。
虚拟筛选可应用于基于结构和基于配体的药物发现。在基于结构的虚拟筛选(SBVS)中,分子对接可根据化合物与靶点位点预测得到的结合亲和力对大型化合物库进行排序。由于需要高通量,对接仅能提供靶点(通常为蛋白质)与配体结合机制的单一快照,从而对结合机制的热力学过程进行了显著近似。另一个方法是分子动力学(MD),其能够对蛋白质与配体相互作用的动态过程进行建模。然而,MD模拟需要大量的时间和计算资源,才能恰当地采样结合过程的自由能面,从而限制了其适用性。另一方面,基于配体的虚拟筛选(LBVS)通常用于能够从已有小分子生物活性数据中获取生物学相关信息的情形,此时不需要已知靶点结构,而是依赖于生物活性预测模型的训练与应用。
随着机器学习(ML)时代的到来,SBVS和LBVS技术正在快速发展。随着近年来包含数十亿个合成可行分子的超大型化合物库的出现,传统的虚拟筛选在计算效率和准确性方面面临着越来越大的挑战。为应对这些挑战,亟需新型的计算方法,能够以成本高效而又稳健的方式进入可触及的化学空间,并结合多种类型的机器学习方法,尤其是近年来快速发展的深度学习(DL)。
AI在基于配体的虚拟筛选的应用
近年来,基于AI的QSAR模型已在许多LBVS实践中成功应用。通过学习2D或3D分子描述符的分子信息来提升预测性能(图1)。常用的LBVS方法在表1中进行了总结。其中一个方法是PyRMD,它使用随机矩阵判别算法,可在有限计算资源下,仅需数小时就能从超大型化合物库中分类活性和非活性分子。QSAR与AI的互补性体现在多种模型的整合中,例如随机森林(RF)、朴素贝叶斯(NB)和支持向量机(SVM)。Gomes等人提出了一种共识QSAR模型,整合了多种机器学习算法,用于发现抗结核化合物,并证明其对耐药菌株表现出较强活性。
另一种LBVS策略是利用AI通过计算与已知活性化合物的结构相似性确定潜在活性分子。Altalib和Salim利用孪生深度学习(Siamese DL)模型来提升相似性搜索的性能。该算法优于传统的Tanimoto系数以及其他方法,尤其在处理异质性数据集时表现突出。
近年来,深度学习(DL)架构已被广泛应用于LBVS模型中。图神经网络(GNNs)因其能够从图结构数据(如化学结构)中学习而成为研究热点。值得注意的是,GNNs主要被用于新型抗生素的发现。例如,Stokes等人使用定向消息传递神经网络(D-MPNNs)识别多种病原体的广谱抑制剂,最终发现了8种在结构上与已知抗生素不同的分子,这些分子对至少一种病原体表现出抑制作用。更为显著的是,其中两种化合物展现出强大的广谱活性,提供了使用DL提升LBVS效能的首批有力证据。Swanson等人提出了SynthMol,这是一种生成模型,能够利用蒙特卡洛树搜索(MCTS)从近300亿个可合成的化学空间中设计新分子。作者通过在鲍曼不动杆菌中应用该模型,成功发现了新型的实验验证抑制剂,从而展示了SynthMol的潜力。
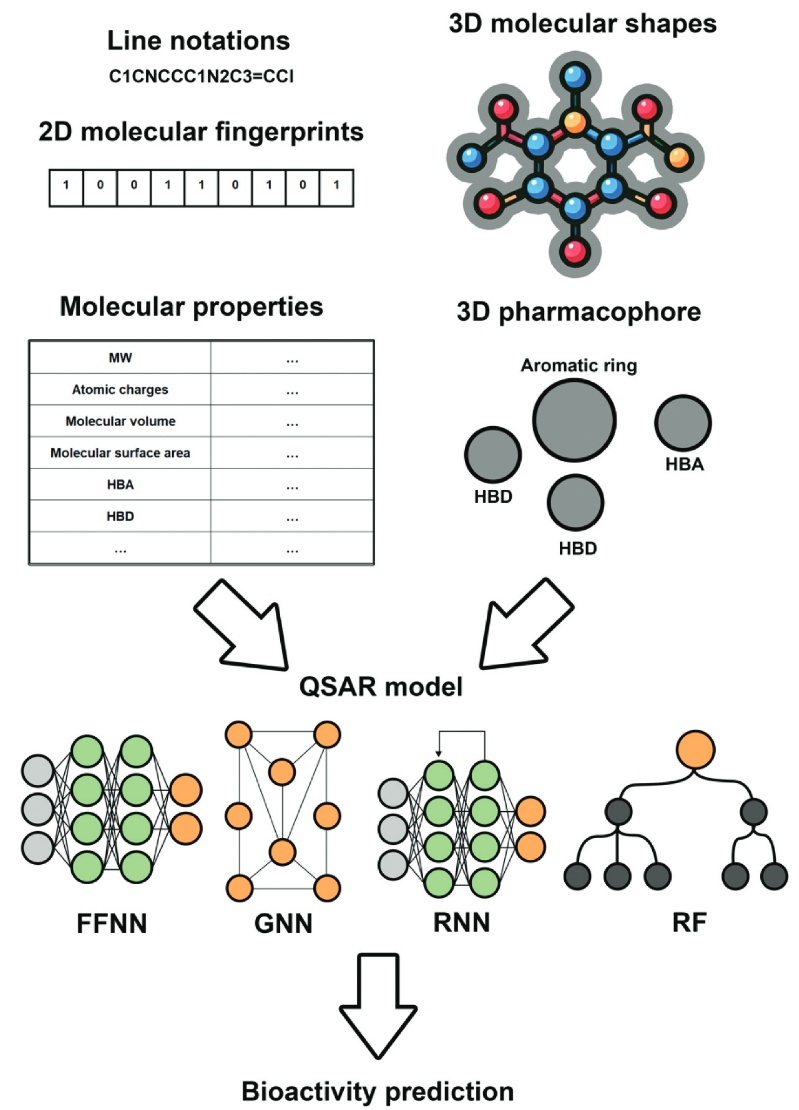
图1 AI在LBVS的应用
尽管取得了成功,LBVS仍面临诸多挑战。其中一个主要限制是泛化能力不足。为应对泛化问题,MoleculeNet框架提出基于分子骨架的训练集、验证集与测试集划分,从而避免不同集合间的重叠。Tossou等人根据训练集-测试集与部署集-训练集间的最小分布差异来选择划分方式,用于研究分子分布外的泛化能力。另一个显著限制是数据稀缺性与数据质量不足。许多公开数据库(如ChEMBL)本质上含有噪声且数据不一致,这在整合不同文献来源的生物活性数据时尤其棘手。为缓解该问题,Landrum和Riniker提出了最大化数据整理方案。
此外,基于配体的方法在中低数据场景下表现有限。为此,主动学习(AL)策略被提出,通过迭代选择小批量样本进行标注并加入训练集,以用更少的数据开发高精度模型。近期,Van Tilborg和Grisoni模拟了一个现实的低数据情景:仅使用1k个分子迭代训练模型,对规模大100倍的化合物库进行虚拟筛选。结果显示,结合AL的方法在多种模型中均优于传统训练。
表1 常用的LBVS方法

最后,AI驱动的LBVS方法评估应遵循标准化指南,以确保不同方法间的公平比较。Wu等人建议使用ROC曲线下面积(AUROC)评估分类器,使用均方根误差(RMSE)评估回归模型。尽管 AUROC相比准确率更稳健,但在分子数据高度不平衡的现实药物发现中可能缺乏意义。精确率-召回曲线下面积(AUPRC)被提出作为替代指标。
尽管存在挑战,AI在LBVS中的作用仍被普遍看好。未来的发展关键在于针对低数据情境的策略(如主动学习与迁移学习)以及生物活性数据的整理与标准化。此外,ML与DL的使用门槛已大幅降低,随着更多用户友好型工具包的出现,这些方法将在未来更加普及,并推动更多AI驱动的LBVS前瞻性验证。
AI在基于结构的虚拟筛选的应用
分子对接
在SBVS中,分子对接是应用最广泛的方法,其目标是预测潜在的蛋白-配体相互作用,并通过打分函数(SFs)对所得结合构象进行排序,以估算结合亲和力。然而,分子对接仍面临重要局限。其一,打分函数与优化/搜索算法之间的平衡是关键挑战。高效的对接不仅需要准确、快速的打分函数计算,还需要稳健的搜索与优化策略,以探索小分子结合于靶点时的构象空间。另一大局限在于通常忽略了蛋白质的柔性,而这对模拟真实的分子相互作用至关重要。为克服这些挑战,近年来提出了多种AI方法以改进分子对接(图2及表2)。
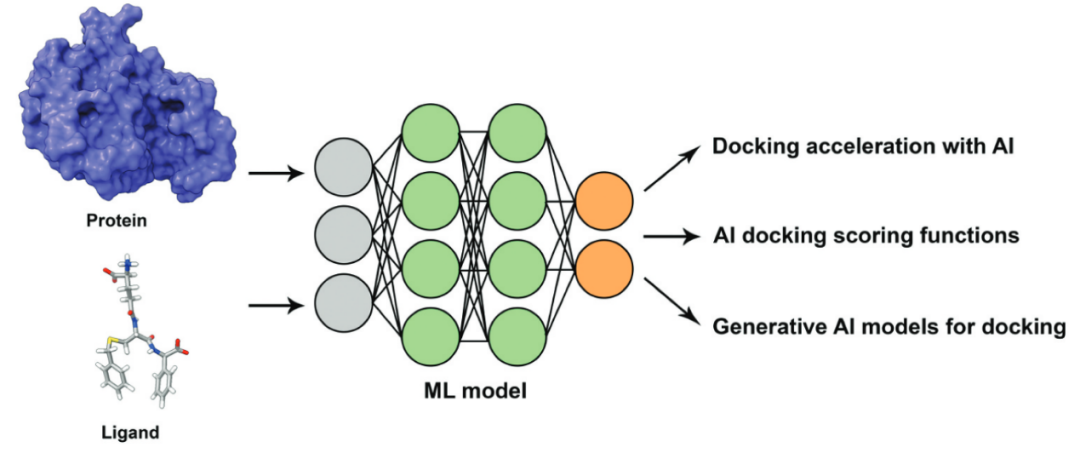
图2 AI在分子对接中的应用
AI在分子对接中的应用主要包括三类方法。其一,使用ML模型加速分子对接,通过训练对接得分的代理模型快速筛选大型化合物库。代表性方法包括Deep Docking、RosettaVs等。另一个活跃的研究方向是开发基于AI的打分函数,以更准确地预测对接中的结合亲和力。代表性方法包括Gnina、EquiScore、PIGNet以及DockBox2。最后一类方法为提出基于生成模型的策略,作为物理采样的替代,用于探索并能量评估广泛的蛋白-配体构象空间,从而更准确地产生合理的结合构象。代表性方法包括DiffDock、SurfDock、KarmaDock等。
表2 用于增强SBVS的AI方法

分子动力学模拟
分子动力学模拟的一个常见应用是估算由对接或生成建模筛选出的候选小分子的相对或绝对结合亲和力,以期提供更可靠的排序并剔除伪活性分子。然而,尽管近年来高性能计算以及GPU加速模拟算法取得了长足进展,MD仍然是一种耗时的技术,尤其在大规模SBVS工作流程中需要消耗大量计算资源。
将AI方法应用于MD模拟仍处于早期阶段,目前主动学习(AL)扮演着最重要的角色。AI有望显著提升MD的计算效率,并改进蛋白-配体结合能的计算(图3)。潜在应用包括基于深度学习的增强采样MD模拟、基于深度学习的模拟分析、以及基于机器学习/深度学习的力场开发(如网络势能模型)。
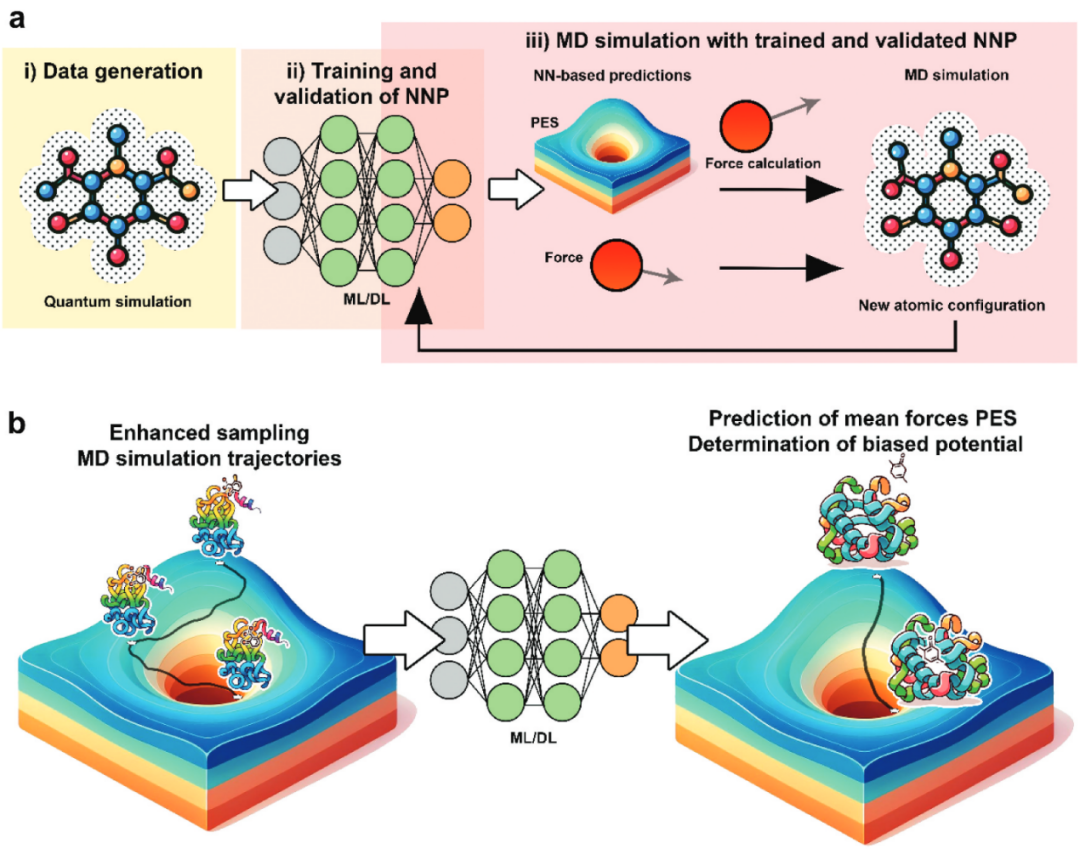
图3 AI在SBVS的MD模拟中的应用
在神经网络势能(NNPs)中,使用具有可训练参数的自适应神经函数。这些参数通过机器学习算法进行优化,以准确模拟原子间相互作用。图3a展示了基于NNPs的MD模拟工作流程。采用量子模拟方法生成原子构型及其对应的力和势能数据集,随后训练并验证模型,使其能够准确预测力和势能面(PES)。将训练好的模型部署在MD模拟中。在每一步模拟中,当前原子构型通过描述符表示后输入训练模型,由模型预测作用在原子上的力,再用这些力更新原子位置以进行下一步模拟。
将AI整合进增强采样MD方法的核心思想在于以更低的成本高效探索复杂分子体系的高维构象空间,从而优于纯物理学方法。为此,已有研究提出利用AI方法,通过增强采样技术衍生的偏置势和集体变量(CVs)来探索复杂的配体-受体势能面(图3b)。这种方法能为虚拟筛选提供显著优势,通过更准确地估计配体结合与动力学参数,来加速新药候选分子的发现。
挑战
在虚拟筛选中,AI学习小分子潜在生物效应背后的化学规律的能力仍存在争议,因为AI预测的分子往往与训练集中的活性化合物存在显著重叠。此外,常用的评估方法(如骨架划分)是否能真实反映实际筛选场景受到质疑,缺乏统一的标准化评估协议。另一大局限是缺乏客观且现实的前瞻性验证。大多数研究仅停留在回顾性或选择性验证,实验验证往往只针对简单靶点,容易产生过于乐观的结果。CACHE挑战公共评测显示,AI方法并未表现出对基于物理的方法的明显优势。因此,作者强烈支持开展无偏基准挑战,用于验证分子构象预测与活性预测模型,相比开发大量复杂但缺乏验证的方法,这种方式对推动该领域发展更具意义。
结语
近来的化学领域进展催生了庞大的化学空间,涵盖数十亿种分子,这促使制药公司和学术机构愈发重视对这一尚未开发的巨大资源进行探索和利用,以寻找新的药物先导化合物。这一趋势推动了AI成为虚拟筛选的核心组成部分,展现出其在加速新型治疗药物发现方面的巨大潜力。因此,将深度学习、主动学习和生成模型与分子对接、分子动力学模拟等传统计算化学方法相结合的新算法与策略,正在学术界与工业界快速发展。然而,尽管取得了显著进展,仍存在诸多挑战,并不断涌现新的问题。因此,研究人员不应因AI的巨大声势与广泛关注而忽视对这些问题的深入解决。
参考链接:
https://doi.org/10.1080/17460441.2025.2508866
--------- End ---------
内容中包含的图片若涉及版权问题,请及时与我们联系删除


评论
沙发等你来抢